Introduction
The phosphoinositide 3-kinase (PI3K) signaling
pathway regulates a multitude of cellular processes including cell
survival, proliferation, migration and invasion. Deregulation of
the PI3K signaling pathway is often detected in various types of
human cancer. The serine/threonine kinase Akt, also known as
protein kinase B (PKB), which was initially identified as a
proto-oncogene Akt8, from a spontaneous thymoma of an AKR mouse
(1), has a strong oncogenic
function and is the primary downstream mediator of PI3K pathway
function (2). In mammals, there are
three different isoforms of Akt, termed Akt1, Akt2 and Akt3, which
share a high degree of homology, while the three Akt isoforms have
some different functions (3).
Amplification of Akt1 was first discovered in a
primary human gastric adenocarcinoma (1). Currently, increasing evidence
indicates that Akt1 has been implicated in the control of various
biological processes, including cell proliferation, survival and
tumor formation. Menges et al (4) demonstrated that Akt1 is positively
correlated with tumor cell proliferation and survival.
Downregulation of Akt1 expression inhibits K562 cell proliferation
(5). Furthermore, by using
bitransgenic MMTV-c-ErbB2, MMTV-myr-Akt1 mouse models,
constitutively active Akt1 markedly accelerated MMTV-c-ErbB2
mammary tumorigenesis (6). However,
the role of Akt1 in cell proliferation remains controversial. For
instance, a recent article indicated that constitutive Akt1 signals
attenuate B-cell receptor signaling and proliferation (7). Therefore, it is essential to
investigate whether ectopic expression of Akt1 promotes cell
proliferation or not in other cell types, including hepatocellular
carcinoma (HCC) and colorectal cancer cells.
Cell migration and invasion are two of the most
important steps involved in cancer metastasis, which accounts for
>90% of all cancer-related deaths (8,9).
Growing evidence indicates that activation of Akt1 is associated
with cancer cell migration, invasion and metastasis. For example,
selective activation of Akt1 not only positively regulates
IGF-1-induced SKOV-3 cell migration and invasion, but also markedly
promotes tumor metastasis (10).
Previous studies showed that Akt isoform Akt1 limits breast cancer
cell motility and invasion (11,12)
and several other independent studies have validated and extended
these observations (13,14). Moreover, these data have been
validated in transgenic mouse models, in which AKT1 was shown to
have an inhibitory effect on breast cancer invasiveness (15,16).
Although overwhelming evidence has demonstrated that the PI3K/Akt
signaling cascade plays a crucial role in the regulation of the
malignant behaviors, including migration and invasion in HCC and
colorectal carcinomas, the role of Akt1 and its precise molecular
mechanisms in these cancer cells remain largely unknown.
In the present study, we demonstrated that
upregulation of Akt1 significantly increased cell proliferation and
enhanced the ability to form colonies in both HepG2 and HCT 116
cells. Furthermore, we also discovered that enforced expression of
Akt1 significantly enhanced the ability of migration and invasion
in HepG2 cells, while it reduced HCT 116 cell migration and
invasion. These data demonstrate a dual role for Akt1 in tumor cell
migration and invasion and highlight the cell type-specific actions
of Akt1 kinases in the regulation of cell motility.
Materials and methods
Materials
3-(4, 5-dimethylthiazol)-2,5-diphenyltetrazolium
bromide (MTT), G418 and wortmannin were purchased from Sigma (St.
Louis, MO, USA). RPMI-1640 and fetal calf serum (FCS) were
purchased from Gibco (Grand Island, NY, USA). The sources of
primary antibodies used for western blot analysis, Akt1, Bcl-2,
NF-κB, MMP2, MMP9, HIF1α, VEGF, as well as horseradish
peroxidase-conjugated anti-mouse and anti-rabbit antibodies were
all purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
All other chemicals used in the experiments were commercial
products of reagent grade.
Cell culture and treatment
Cell lines from human HCC (HepG2) and colorectal
carcinoma (HCT 116) were purchased from the Cell Bank of the
Chinese Academy of Science (Shanghai, China). These cells were
maintained in RPMI-1640 medium supplemented with 10% FCS, 100 U/ml
penicillin, and 100 μg/ml streptomycin at 37°C in a humidified
incubator containing 5% CO2.
Plasmid construction and
transfection
The full-length coding region (1443 bp) of Akt1 was
amplified from human genomic DNA by reverse
transcription-polymerase chain reaction (RT-PCR) using the
following primers: Akt1-sense 5′-CCG CTC GAG ACC ATG AGC GAC GTG
GCT ATT GTG AAG-3′, and antisense 5′-CGG GAT CCG CAG TCC ACC GCC
GCC TCAG-3′, and the PCR products were then digested with
XhoI/BamHI and were inserted into the pIRES2-EGFP
vector (Clontech Laboratories, Inc., Palo Alto, CA, USA) carrying
neomycin resistance gene. The recombinant construct was verified by
direct DNA sequencing.
For transfection, HepG2 and HCT 116 cells were
seeded in 24-well plates at 5×104/well and incubated at
37°C in a humidified incubator containing 5% CO2. The
next day, when these cells were ~80% confluent, cells were
transfected with TurboFect™ in vitro transfection reagent
according to the manufacturer’s instructions. Briefly, recombinant
plasmid (1 μg) was mixed with TurboFect™ reagent and pre-incubated
for 20 min at room temperature in 100 μl of serum-free RPMI-1640.
HepG2 and HCT 116 cells transfected with pIRES2-EGFP-Akt1 and
pIRES2-EGFP vector were termed as HepG2-Akt1 and HepG2-Vec, HCT
116-Akt1 and HCT 116-Vec, respectively. These transfected cells
were selected in the presence of G418 (100 μg/ml) for 2 weeks, and
then the stable plasmid-transfected clones were generated by using
limiting dilution analyses in 96-well plates.
Cell growth curve analysis
The MTT assay was used to detect the proliferation
rate of cells transfected with empty vector or Akt1 plasmid. The
process was performed as previously described with some
modifications (17). Briefly, 2,000
cells/well were plated in 96-well plates and incubated for 1, 2, 3,
4, 5, 6 and 7 days, respectively. Then, 50 μl MTT reagents (1
mg/ml) was added at indicated time-points and cells were incubated
for 4 h at 37°C. Supernatants were removed from the wells and 100
μl DMSO was pipetted to solubilize the crystal product for 10 min
at room temperature. The absorbance (OD) of each well was measured
with a microplate reader (Bio-Rad Laboratories) at a wavelength of
570 nm.
RNA isolation and RT-PCR
Total RNA was extracted using TRIzol reagent
(Invitrogen). Briefly, 2 μg of total RNA was subjected to DNase I
digestion (1 U/μl; Fermentas, Hanover, MD, USA) at 37°C for 30 min
and then to heat inactivation of DNase I at 70°C for 15 min,
followed by reverse-transcription using Moloney murine leukemia
virus reverse transcriptase (Promega). The PCR primers and regimen
were: 5′-ATG AGC GAC GTG GCT ATT GTG AAG-3′ and 5′-GAG GCC GTC AGC
CAC AGT CTG GATG-3′ for Akt1 (330 bp); 5′-CGG AGT CAA CGG ATT GGT
CGT AT-3′, and 5′-AGC CTT CTC CAT GGT GG TGA AGAC-3′ for GAPDH (307
bp). All PCR reactions were performed using standard PCR
conditions: 95°C for 5 min, 95°C for 45 sec, annealing at different
temperatures for each gene respectively for 45 sec, extension at
72°C for 1 min for 30 cycles, and a final extension at 72°C for 10
min. PCR products were separated on 1.0% agarose gel. The gel was
then digitally photographed and scanned with UVI Gel Analyzing
System (UVI Tech, Cambridge, UK).
Colony formation assay
Cells were plated in a fresh 24-well plate at a
density of 200 cells/well and maintained in RPMI-1640 containing
10% FCS. The medium was changed every 3 days for 14 days until
visible colonies formed. Colonies were fixed and stained with 0.1%
crystal violet in 20% methanol for 15 min. Individually stained
colonies (>50 cells) were counted in each well. The colony
formation assay was performed as previously reported with some
modifications (18).
Wound scratch assay
The wound scratch assay was performed as previously
described (19). Briefly, cells
were grown to confluence overnight prior to serum starvation for 24
h. The confluent cell monolayer was then scratched with a pipette
tip (20 μl) and washed thrice with PBS to remove floating cells.
After the line scratch, the width of the wound was measured and
recorded as t=0. The cells were then allowed to migrate back into
the wounded area and the closing of the wound was measured at 24 h.
The migration distance (in μm) was determined as the reduction of
the width of the open area.
Boyden chamber assay
The migration and invasion assays were performed in
24-well Boyden chambers with 8 μm pore size polycarbonate membranes
(Corning, Corning, NY, USA), as previously described (20). For the invasion assay, the membrane
was coated with 15 μg Matrigel (R&D Systems) to form a matrix
barrier. Serum-starved cells (1×105 cells) were seeded
into the upper compartment of the chamber in serum-free medium,
supplemented with 100 nM wortmannin or not, while the lower
compartment was filled with 600 μl of DMEM containing 10% FBS.
Following incubation at 37°C for 24 h, tumor cells remaining on the
upper surface of the membrane were removed with cotton swabs. The
cells on the lower surface of the membrane were fixed, stained with
crystal violet and then counted under a light microscope. For the
migration assay, only one third of cells was applied to the
Transwell chamber without Matrigel.
Western blot analysis
After treatment with 100 nM wortmannin or not for 24
h, cells were harvested and washed with PBS. Cell lysates were
prepared in the protein extraction buffer containing 150 mM NaCl,
10 mM Tris (pH 7.2), 5 mM EDTA, 0.1% Triton X-100, 5% glycerol and
2% SDS. Western blot analysis was performed as previously described
(21). The total protein
concentration was determined using the protein assay kit (Beyotime,
China). Cell lysates in 5X SDS-sample buffer were boiled for 5 min
and then equal amounts of total proteins were separated using 10 or
12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF)
membranes (Immobilion Millipore). Membranes were then blocked in
PBST containing 5% dried skimmed milk for 1 h at room temperature.
The blots were incubated with corresponding primary antibodies at
4°C overnight. After washing three times with PBST, the membranes
were incubated with corresponding horseradish peroxidase-conjugated
goat anti-mouse or anti-rabbit secondary antibody, and then washed
three times with PBST. Proteins were detected using the ECL plus
reagents (Beyotime, China).
Statistical analysis
Data are expressed as the means ± SD from at least
three independent experiments. Unless otherwise noted, the
differences between groups were assessed by ANOVA. All the tests
performed in the present study were two-sided using SPSS 13.0
software (SPSS Inc., Chicago, IL, USA). Differences were considered
statistically significant at values of P<0.05.
Results
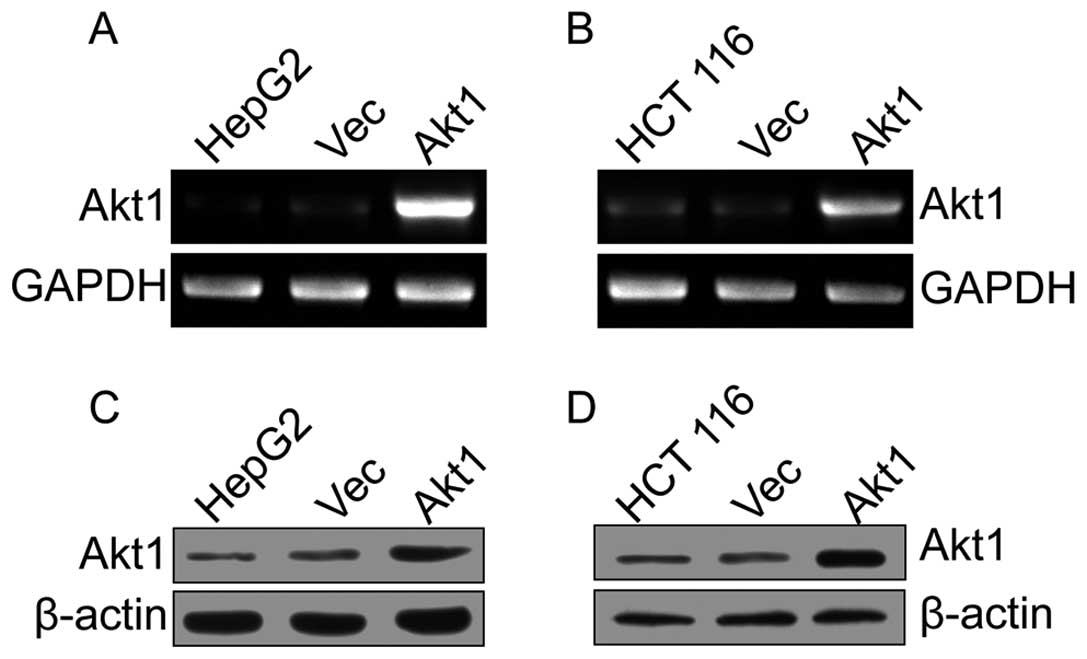
Akt1 is stably overexpressed in HepG2 and
HCT 116 cells
After stably transfected cells were individually
selected, the Akt1 and GAPDH mRNA levels were measured using
RT-PCR. Compared with the control group, the pIRES2-EGFP-Akt1
transcripts were strongly upregulated (Fig. 1A and B). Consistent with our RT-PCR
data, western blot analysis revealed that Akt1 protein levels were
also clearly increased in pIRES2-EGFP-Akt1-transfected cells
(Fig. 1C and D). The pIRES2-EGFP
empty vector did not substantially affect the endogenous Akt1
expression in both mRNA and protein levels. Collectively, these
findings indicate that Akt1 is stably overexpressed in HepG2 and
HCT 116 cells.
Akt1 upregulation promotes cell
proliferation and colony formation in HepG2 and HCT 116 cells
We then determined the functional consequences of
Akt1. As shown in Fig. 2A and B,
compared with the controls, the proliferation rates of HepG2-Akt1
and HCT 116-Akt1-transfected cells were increased significantly
(P<0.01); then, the capacity of colony formation was evaluated
on these cell lines. Our results showed that the number of colonies
of HepG2-Akt1 and HCT 116-Akt1 cells were 66.7±9.6 and 71.7±5.9,
respectively; however, the number of colonies was only 29.3±4.7 and
29.0±6.0 for HepG2-Vec and HCT 116-Vec cells, respectively. There
was a statistically significant increase in the number of colonies
of HepG2-Akt1 and HCT 116-Akt1 cells compared to that of the
individual control groups (Fig. 2C and
D; P<0.01, P<0.001), while wortmannin significantly
attenuated the colonies of the tumor cells with upregulation of
Akt1. These results indicate that Akt1 promotes tumor cell
proliferation and colony formation.
Upregulation of Akt1 expression promotes
HepG2 cell migration and invasion
Next, we examined the effect of Akt1 on HepG2 cell
migration and invasion. As shown in Fig. 3A, a significant difference between
wound distance of HepG2-Akt1 cells compared to HepG2-Vec cells was
observed. Forced expression of Akt1 markedly stimulated wound
closure compared with the empty vector-transfected cells, while
this effect was significantly decreased after treatment with
wortmannin (Fig. 3A). Boyden
chamber Transwell assay also showed that the migration of HepG2
cells was increased by >2-fold due to Akt1 overexpression, but
was significantly decreased when treated with wortmannin (Fig. 3C).
To determine whether Akt1 also regulates HepG2 cell
invasion, the Transwell assay was repeated. As shown in Fig. 4A, upregulation of Akt1 expression
markedly induced cell invasion compared with the control group,
whereas cell invasion was strongly suppressed by wortmannin. Taken
together, these results demonstrate that Akt1 is correlated with
cell migration and invasion potential of liver cancer HepG2
cells.
Overexpression of Akt1 suppresses the
migration and invasion of HCT 116 cells
We determined the role of Akt1 in HCT 116 cell
migration. Upregulation of Akt1 expression markedly suppressed
wound closure compared with HCT 116-Vec cells, while wortmannin
treatment significantly reversed the effects of Akt1 on HCT 116
cells (Fig. 3B). The Transwell
assay also indicated that ectopic expression of Akt1 noticeably
decreased HCT 116-Akt1 cell migration by ~50% in comparison with
the control group (Fig. 3D). The
addition of wortmannin significantly attenuated the inhibitory
effect of Akt1 on HCT 116 cell migration (Fig. 3D). We then examined whether Akt1
also regulates HCT 116 cell invasion. Akt1 overexpression
significantly inhibited invasion compared to control cells
transfected with empty vector (Fig.
4B), while wortmannin partially antagonized the inhibitory
effect of Akt1 on migration and invasion of HCT 116 cells (Fig. 4B). These findings suggest that Akt1
suppresses the cell migration and invasion of HCT 116 cells.
Cellular signaling pathways involved in
Akt1 action
To identify the cellular signaling pathways involved
in Akt1 action, we first examined the pro-survival and
proliferative molecules downstream of Akt1, including Bcl-2 and
nuclear factor-κB (NF-κB). Overexpression of Akt1 significantly
induced the expression of Bcl-2 and NF-κB in both HepG2 (Fig. 5A) and HCT 116 cells (Fig. 5B). However, wortmannin partly
reversed Akt1-mediated effects.
Overexpression of MMPs is associated with tumor
invasion and metastasis and, in particular, MMP2 and MMP9, which
are well known to play a pivotal role in tumor invasion and
metastasis development in various types of cancer including
hepatocellular and colorectal carcinomas (22,23).
Our results also showed that Akt1 overexpression significantly
increased the expression of MMP2 and MMP9 in HepG2 cells, while
there was an opposing effect of Akt1 in HCT 116 cells. Of note,
wortmannin significantly attenuated these effects of Akt1 in both
types of tumor cells (Fig. 5). Our
results also demonstrated that upregulation of Akt1 also enhanced
the expression of HIF-1α and VEGF in HepG2 cells, while it
attenuated the levels of these two types of proteins in HCT 116
cells.
Discussion
Activation of the PI3K/Akt signaling pathway has
been detected in several types of cancer, and PI3K or its
downstream components, including AKTs, are considered attractive
targets for cancer therapy. However, several studies have
highlighted that the biological outcomes obtained upon AKT
inhibition are very complex (13,24,25),
including the potential cell-type specific effects of AKT isoform
Akt1 on cell migration and invasion (10). Therefore, it is necessary to
investigate the potential role of Akt1 in various types of human
cancer before its inhibitors are used as cancer therapies.
In the present study, our findings showed that
upregulation of Akt1 resulted in increased cell proliferation in
both HepG2 and HCT 116 cells. Inhibition of PI3K by wortmannin
efficiently reduced the colonies in tumor cells with Akt1
transfection. Consistent with our findings, ablation of AKT1
decreased the proliferation of the androgen-independent cell line
PC-3 (26). Moreover, as these are
in vitro assays, validation of these results in vivo
is required and only few reports have already addressed these
issues. Using mice bitransgenic of mammary tumorigenesis with
constitutively active Akt1 and ErbB-2, Young et al (6) reported that overexpression of
activated Akt1 resulted in increased tumor frequency. In contrast,
loss of Akt1 reduced tumor proliferative function in a murine model
of thyroid cancer (27). These
results suggested that Akt1 expression in tumor cells is required
for cell proliferation, although how ectopic expression of Akt1
promotes tumor cell proliferation remains unclear.
We investigated the potential cellular signaling
pathway involved in Akt1-induced cell proliferation. Bcl-2 is a
well-known pro-survival gene downstream of Akt1. It is often
overexpressed in various human tumors. In the present study, our
results indicated that Bcl-2 is overexpressed in both HepG2-Akt1
and HCT 116-Akt1 cells compared with their respective
vector-transfected cells. Upon wortmannin treatment, the expression
of Bcl-2 was significantly downregulated, suggesting that Bcl-2 may
play a key role in Akt1-induced proliferation. In concordance with
our observations, a recent study indicated that the Akt1/Bcl-2
signaling pathway plays a critical role in
angiopoietin-2-stimulated cell survival and proliferation both
in vitro and in vivo (28). Moreover, silencing of Akt1 was
associated with the suppression of Bcl-2 which, in turn, resulted
in the inhibition of cell proliferation, stimulation of apoptosis
in vitro and inhibition of xenograft growth in nude mice
(29).
Nuclear factor-κB (NF-κB) is also a ubiquitous
transcription factor. It is often thought to contribute to
malignant transformation by aberrant activation of cellular
functions that are commonly associated with tumor promotion,
including stimulating cell growth and proliferation (30). Furthermore, Han et al
(31) supported the notion that
constitutive activation of the PI3K/Akt1/NF-κB signaling pathway is
important for cell survival and proliferation in iMycEμ
B-cell lymphomas. In the present study, enforced expression of Akt1
increased the expression of NF-κB in both HepG2 and HCT 116 cells.
Upon wortmannin treatment, the level of NF-κB was markedly
downregulated comparable to that of HepG2-Vec and HCT 116-Vec
cells, respectively. Considering that, the expression of Bcl-2 and
NF-κB is in concordance with Akt1-mediated cell proliferation in
both HepG2 and HCT 116 cells. Hence, we proposed that Bcl-2 and
NF-κB are two important molecules involved in Akt1-regulated cell
proliferation.
In addition to the effect of Akt1 on tumor cell
growth, its effects on tumor cell migration and invasion are of
considerable importance. In the present study, we demonstrated that
upregulation of Akt1 expression in liver cancer HepG2 cells
markedly promoted the wound closure and the number of migrated
cells. Moreover, enforced expression of Akt1 also significantly
enhanced the invasion of HepG2 cells. Treatment with wortmannin
significantly attenuated the positive effect of Akt1 on HepG2 cell
migration and invasion. Then, we examined whether Akt1 plays a
similar role in HCT 116 cells. Notably, the results from the wound
healing assay suggested that overexpression of Akt1 significantly
inhibited the wound closure of HCT 116 cells, while wortmannin
partially rescued the inhibitory effect of Akt1 on colorectal tumor
HCT 116 cells. In addition, the results from the Transwell
migration assay also verified that enforced Akt1 expression
significantly reduced HCT 116 cell migration compared with the
corresponding control group. Furthermore, we also discovered that
upregulation of Akt1 expression significantly inhibited HCT 116
cell invasion. However, following treatment with wortmannin, the
inhibitory effect of Akt1 on HCT 116 cell migration and invasion
was considerably alleviated. Considering the completely inverse
effect of Akt1 on the migration and invasion in HepG2 and HCT 116
cells, we propose that the potential molecular mechanisms may be
different in these two types of cell lines.
Matrix metalloproteinases (MMPs) are a large family
of enzymes, which play a fundamental role in various components of
the extracellular matrix degradation and remodeling (32). MMPs have been found to be
overexpressed in several human tumors and correlate with advanced
stage, invasion, metastatic properties and poor prognosis (33). Among secreted MMPs, MMP2 and MMP9
are well known to play a pivotal role in tumor invasion and
metastasis development in several types of cancer including
hepatocellular and colorectal carcinomas (22,23).
Moreover, it has been shown that silencing of Akt1 is associated
with reduced expression of MMP2 and MMP9 in both SGC7901 gastric
adenocarcinoma and U251 glioma cells (34). Consistent with these reports, our
results also showed that upregulation of Akt1 resulted in increased
expression of MMP2 and MMP9 in HepG2 cells, while it inhibited the
expression of MMP2 and MMP9 in colorectal carcinoma HCT 116 cells.
Treatment with pharmacologic inhibitor of PI3K completely reversed
the effect of Akt1 in both cell types. In addition, the protein
levels of HIF1α and VEGF were also significantly increased in
HepG2-Akt1 cells compared to its control; by contrast, they were
degraded in HCT 116 cells. Upon wortmannin treatment, the
expression of HIF1α and VEGF was strongly suppressed in HepG2
cells, whereas it was upregulated in HCT 116 cells. Consistent with
these results, activation of the PI3K/Akt1 pathway in tumor cells
resulted in increased VEGF secretion, both by HIF-1α-dependent and
-independent mechanisms. Moreover, sustained endothelial activation
of Akt1 has been shown to induce the formation of structurally
abnormal blood vessels (35).
Furthermore, silencing of Akt1 by siRNA markedly decreased HIF-1α
translation in normoxia in the presence of dimethyloxallyl glycine
and in hypoxia (36). Collectively,
these findings suggested that several signaling molecules,
including MMP2, MMP9, HIF1α and VEGF, may play a key role in
Akt1-mediated cell migration and invasion. Meanwhile, we proposed
that Akt1 upregulation may first promote the expression of MMP2,
MMP9, HIF1α and VEGF, which contributed to inducing the migration
and invasion of HepG2 cells. In contrast, in colorectal carcinoma
HCT 116 cells, upregulation of Akt1 suppressed the expression of
these genes, which resulted in reduced cell migration and invasion.
However, how Akt1 regulates these multiple molecules remains to be
further investigated.
In the present study, we demonstrated that
upregulation of Akt1 promotes cell proliferation in both HepG2 and
HCT 116 cells, while the role of Akt1 in cell migration and
invasion is completely distinct in these two cell types, suggesting
the role of Akt1 in cell proliferation is independent of cell
migration and invasion, although all are malignant phenotypes in
the process of tumor development. Consistent with our observations,
Pierau et al (7)
demonstrated that constitutive Akt1 signals attenuate B-cell
receptor signaling and proliferation, but enhance B-cell migration
and effector function. Moreover, our results also demonstrated that
overexpression of Akt1 promoted the cell migration and invasion of
human liver cancer HepG2 cells; however, it attenuated the cell
migration and invasion of human colorectal carcinoma HCT 116 cells.
Therefore, we proposed that the effect of Akt1 on cell migration
and invasion is likely due to the difference in cell type and
context. Indeed, it has been shown that human non-small cell lung
cancer (NSCLC) A549 cells with an expression construct for Akt1,
exhibited significantly higher invasive ability through Matrigel
than those cells with a control empty vector (37). Knockdown of Akt1 expression in NSCLC
cells resulted in decreased cell migration (38). Furthermore, high levels of total
Akt1 were also associated with increased lymph node metastasis in
human prostate cancer (39). In the
present study, ectopic expression of Akt1 significantly promoted
the cell proliferation, migration and invasion in human liver
cancer HepG2 cells, suggesting that Akt1 is may be a promising
target for human liver cancer therapy.
On the other hand, Yoeli-Lerner et al
(12) revealed that expression of
activated Akt1 potently blocked the cell migration and invasion
through Matrigel in three distinct breast cancer cell lines. Irie
et al (13) also discovered
that silencing of Akt1 expression markedly enhanced cell migration
induced by growth factor in MCF-10A cells. Consistent with these
observations, in the present study, upregulation of Akt1 expression
significantly inhibited the cell migration and invasion in
colorectal carcinoma HCT 116 cells. Furthermore, in vivo
evidence also identified the role for Akt1 in blunting breast
cancer cell invasion and subsequent metastasis. Using a mouse
bitransgenic assay of mammary tumorigenesis with constitutively
activated Akt1 and ErbB-2, Hutchinson et al (15) reported that upregulation of
activated Akt1 expression resulted in a decrease in the incidence
of metastatic lesions compared with control animals.
Although we, and other groups, have demonstrated the
inhibitory effect of Akt1 on cell migration and invasion in several
types of tumor cells, the precise cell signaling mechanisms remain
to be further investigated. Multiple molecular mechanisms have been
shown to be involved in Akt1-mediated negative regulation of cell
migration and invasion, including inhibition of NFAT transcription
factor (12) or phosphorylation of
paladin (11), activation of ERK
signaling pathway (13) and cell
surface B1-integrins (14). Of
note, silencing of AKT1 in PC-3 cells also resulted in strong
upregulation of VEGFR2 (14).
Consistently, in the present study, we directly demonstrated that
enforced expression of Akt1 significantly not only reduced the
expression of VEGF, but also suppressed the expression of MMP2,
MMP9 and HIF1α, which then suppressed the migration and invasion of
HCT 116 cells.
In the present study, our experiments on Akt1 were
limited to human hepatocellular carcinoma HepG2 cells and
colorectal carcinoma HCT 116 cells. Enforced Akt1 expression
significantly increased cell proliferation through induction of
Bcl-2 and NF-κB in both HepG2 and HCT 116 cells. Moreover, our
observations also showed that Akt1 overexpression significantly
promoted the expression of MMP2, MMP9, HIF1α and VEGF, which then
contributed to enhancing the migration and invasion of HepG2 cells.
Upregulation of Akt1 expression markedly downregulated the levels
of these proteins and suppressed the migration and invasion of HCT
116 cells. In order to verify these effects of Akt1, more types of
liver cancer and colorectal carcinoma cell lines are required.
Furthermore, the precise molecular mechanisms remain to be further
investigated.
Although Akt1 contributes to proliferation in both
types of tumor cells, specific pharmacological inhibition may have
a differential impact on migration and invasion in different cell
types. Understanding these differences is crucial to the
implication of specific inhibitors for cancer therapies.
Acknowledgements
The present study was supported by the NSFC-Henan
‘Talented Man’ Train Union Fund (no. U1204829) and the Projects of
Science and Technology of Henan (no. 102300410095).
References
|
1
|
Staal SP: Molecular cloning of the akt
oncogene and its human homologues AKT1 and AKT2: amplification of
AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci
USA. 84:5034–5037. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jiang BH, Aoki M, Zheng JZ, Li J and Vogt
PK: Myogenic signaling of phosphatidylinositol 3-kinase requires
the serine-threonine kinase Akt/protein kinase B. Proc Natl Acad
Sci USA. 96:2077–2081. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fernandez-Hernando C, Jozsef L, Jenkins D,
Di Lorenzo A and Sessa WC: Absence of Akt1 reduces vascular smooth
muscle cell migration and survival and induces features of plaque
vulnerability and cardiac dysfunction during atherosclerosis.
Arterioscler Thromb Vasc Biol. 29:2033–2040. 2009. View Article : Google Scholar
|
|
4
|
Menges CW, Sementino E, Talarchek J, et
al: Group I p21-activated kinases (PAKs) promote tumor cell
proliferation and survival through the AKT1 and Raf-MAPK pathways.
Mol Cancer Res. 10:1178–1188. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Okumura N, Yoshida H, Nishimura Y,
Kitagishi Y and Matsuda S: Terpinolene, a component of herbal sage,
downregulates AKT1 expression in K562 cells. Oncol Lett. 3:321–324.
2012.PubMed/NCBI
|
|
6
|
Young CD, Nolte EC, Lewis A, Serkova NJ
and Anderson SM: Activated Akt1 accelerates MMTV-c-ErbB2 mammary
tumourigenesis in mice without activation of ErbB3. Breast Cancer
Res. 10:R702008. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pierau M, Na SY, Simma N, et al:
Constitutive Akt1 signals attenuate B-cell receptor signaling and
proliferation, but enhance B-cell migration and effector function.
Eur J Immunol. 42:3381–3393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sporn MB: The war on cancer. Lancet.
347:1377–1381. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim EK, Yun SJ, Ha JM, et al: Selective
activation of Akt1 by mammalian target of rapamycin complex 2
regulates cancer cell migration, invasion, and metastasis.
Oncogene. 30:2954–2963. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chin YR and Toker A: The actin-bundling
protein palladin is an Akt1-specific substrate that regulates
breast cancer cell migration. Mol Cell. 38:333–344. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoeli-Lerner M, Yiu GK, Rabinovitz I,
Erhardt P, Jauliac S and Toker A: Akt blocks breast cancer cell
motility and invasion through the transcription factor NFAT. Mol
Cell. 20:539–550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Irie HY, Pearline RV, Grueneberg D, et al:
Distinct roles of Akt1 and Akt2 in regulating cell migration and
epithelial-mesenchymal transition. J Cell Biol. 171:1023–1034.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Virtakoivu R, Pellinen T, Rantala JK,
Perala M and Ivaska J: Distinct roles of AKT isoforms in regulating
β1-integrin activity, migration, and invasion in prostate cancer.
Mol Biol Cell. 23:3357–3369. 2012.
|
|
15
|
Hutchinson JN, Jin J, Cardiff RD, Woodgett
JR and Muller WJ: Activation of Akt-1 (PKB-α) can accelerate
ErbB-2-mediated mammary tumorigenesis but suppresses tumor
invasion. Cancer Res. 64:3171–3178. 2004.
|
|
16
|
Maroulakou IG, Oemler W, Naber SP and
Tsichlis PN: Akt1 ablation inhibits, whereas Akt2 ablation
accelerates, the development of mammary adenocarcinomas in mouse
mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T
transgenic mice. Cancer Res. 67:167–177. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xie SQ, Li Q, Zhang YH, et al: NPC-16, a
novel naphthalimide-polyamine conjugate, induced apoptosis and
autophagy in human hepatoma HepG2 cells and Bel-7402 cells.
Apoptosis. 16:27–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu T, Zhu Y, Xiong Y, Ge YY, Yun JP and
Zhuang SM: MicroRNA-195 suppresses tumorigenicity and regulates
G1/S transition of human hepatocellular carcinoma cells.
Hepatology. 50:113–121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Koomoa DL, Geerts D, Lange I, et al:
DFMO/eflornithine inhibits migration and invasion downstream of
MYCN and involves p27Kip1 activity in neuroblastoma. Int
J Oncol. 42:1219–1228. 2013.PubMed/NCBI
|
|
20
|
Fang JH, Zhou HC, Zeng C, et al:
MicroRNA-29b suppresses tumor angiogenesis, invasion, and
metastasis by regulating matrix metalloproteinase 2 expression.
Hepatology. 54:1729–1740. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xie SQ, Zhang YH, Li Q, et al:
3-Nitro-naphthalimide and nitrogen mustard conjugate NNM-25 induces
hepatocellular carcinoma apoptosis via PARP-1/p53 pathway.
Apoptosis. 17:725–734. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li J, Lau GK, Chen L, et al: Interleukin
17A promotes hepatocellular carcinoma metastasis via NF-κB induced
matrix metalloproteinases 2 and 9 expression. PLoS One.
6:e218162011.
|
|
23
|
Yang P, Yuan W, He J, et al:
Overexpression of EphA2, MMP-9, and MVD-CD34 in hepatocellular
carcinoma: implications for tumor progression and prognosis.
Hepatol Res. 39:1169–1177. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chandarlapaty S, Sawai A, Scaltriti M, et
al: AKT inhibition relieves feedback suppression of receptor
tyrosine kinase expression and activity. Cancer Cell. 19:58–71.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dillon RL and Muller WJ: Distinct
biological roles for the akt family in mammary tumor progression.
Cancer Res. 70:4260–4264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cariaga-Martinez AE, Lopez-Ruiz P,
Nombela-Blanco MP, et al: Distinct and specific roles of AKT1 and
AKT2 in androgen-sensitive and androgen-independent prostate cancer
cells. Cell Signal. 25:1586–1597. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Saji M, Narahara K, McCarty SK, et al:
Akt1 deficiency delays tumor progression, vascular invasion, and
distant metastasis in a murine model of thyroid cancer. Oncogene.
30:4307–4315. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Imanishi Y, Hu B, Xiao G, Yao X and Cheng
SY: Angiopoietin-2, an angiogenic regulator, promotes initial
growth and survival of breast cancer metastases to the lung through
the integrin-linked kinase (ILK)-AKT-B cell lymphoma 2 (Bcl-2)
pathway. J Biol Chem. 286:29249–29260. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang L, Xiao L, Ma X, et al: Effect of
DNAzymes targeting Akt1 on cell proliferation and apoptosis in
nasopharyngeal carcinoma. Cancer Biol Ther. 8:366–371. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Basseres DS and Baldwin AS: Nuclear
factor-κB and inhibitor of κB kinase pathways in oncogenic
initiation and progression. Oncogene. 25:6817–6830. 2006.
|
|
31
|
Han SS, Yun H, Son DJ, et al:
NF-κB/STAT3/PI3K signaling crosstalk in iMycEμ B
lymphoma. Mol Cancer. 9:972010.
|
|
32
|
Egeblad M and Werb Z: New functions for
the matrix metalloproteinases in cancer progression. Nat Rev
Cancer. 2:161–174. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rundhaug JE: Matrix metalloproteinases and
angiogenesis. J Cell Mol Med. 9:267–285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang J, Zhang QY, Fu YC, et al:
Expression of p-Akt and COX-2 in gastric adenocarcinomas and
adenovirus mediated Akt1 and COX-2 ShRNA suppresses SGC-7901
gastric adenocarcinoma and U251 glioma cell growth in vitro and in
vivo. Technol Cancer Res Treat. 8:467–478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Karar J and Maity A: PI3K/AKT/mTOR pathway
in angiogenesis. Front Mol Neurosci. 4:512011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pore N, Jiang Z, Shu HK, Bernhard E, Kao
GD and Maity A: Akt1 activation can augment hypoxia-inducible
factor-1α expression by increasing protein translation through a
mammalian target of rapamycin-independent pathway. Mol Cancer Res.
4:471–479. 2006.PubMed/NCBI
|
|
37
|
Lee YC, Lin HH, Hsu CH, Wang CJ, Chiang TA
and Chen JH: Inhibitory effects of andrographolide on migration and
invasion in human non-small cell lung cancer A549 cells via
down-regulation of PI3K/Akt signaling pathway. Eur J Pharmacol.
632:23–32. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee MW, Kim DS, Lee JH, et al: Roles of
AKT1 and AKT2 in non-small cell lung cancer cell survival, growth,
and migration. Cancer Sci. 102:1822–1828. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li R, Dai H, Wheeler TM, et al: Prognostic
value of Akt-1 in human prostate cancer: a computerized
quantitative assessment with quantum dot technology. Clin Cancer
Res. 15:3568–3573. 2009. View Article : Google Scholar : PubMed/NCBI
|