Introduction
Hepatocellular carcinoma (HCC) is the most common
primary form of liver cancer and the third most fatal type of
cancer globally, after lung and stomach cancer (1,2). With
>750,000 new cases diagnosed every year worldwide, HCC is the
sixth most common neoplasm (3). The
overall 5-year survival rate of liver cancer patients remains low,
ranging from 0 to 14% (4). This is
due to the fact that HCC is diagnosed at an advanced/symptomatic
stage in most patients, when limited therapeutic options such as
surgery and topical therapy (including radiofrequency ablation) are
available (5,6). This illustrates the importance of
elucidating the cellular and molecular mechanisms involved in this
aggressive cancer to develop more effective treatment options and
improve the prognosis of HCC patients.
Approximately 90% of HCC cases arise from cirrhosis
and the disease is strongly associated with several risks factors,
including hepatitis B and C infections, alcohol abuse, primary
biliary cirrhosis, autoimmune hepatitis and nonalcoholic
steatohepatitis (7,8). Different players, including immune
cells, hepatic stellate cells, and macrophages, react to liver
injury by producing cytokines and components of the extracellular
matrix, which promote angiogenesis and survival of damaged
hepatocytes or cancer stem cells (9–12).
This regenerative response favors the accumulation of mutations and
epigenetic aberrations, which leads to malignant transformation of
preneoplastic nodules (2,13,14).
However, similar to other types of cancer, the molecular mechanisms
underlying the development and progression of HCC remain
unclear.
DNA methylation is a major epigenetic mechanism of
gene regulation occurring in eukaryotic DNA at CpG sites, which are
generally enriched in the promoters of genes. In a wide range of
tumors, including HCC, global hypomethylation and specific promoter
hypermethylation have been linked with genomic instability and
inactivation of tumor suppressor genes (TSGs), which regulate a
variety of important cellular networks including apoptosis, DNA
repair, inflammation, cell adhesion and migration, as well as
cell-cycle control (14–17). It has been shown that specific drugs
can reverse hypermethylation, and, hence, the application of DNA
demethylation has been investigated to explore its therapeutic
ability to reactivate TSGs whose subsequent gene expression leads
to the inhibition of tumor progression (18,19). A
number of DNA methylation inhibitors are currently under
investigation, including azacytidine and decitabine. Since
epigenetic alterations in cancer cells affect virtually every
cellular pathway such as those involved in cell-cycle progression,
angiogenesis, apoptosis, cell survival and immunogenicity, it is
thought that epigenetic drugs will possess versatile antitumor
activity (18,20,21).
In addition, abnormal hypermethylated genes in cancer can serve as
biomarkers for early detection and tumor classification, and for
monitoring response to treatments such as targeted therapy and
epigenetic agents (22–24).
The tumor suppressor candidate 1 (TUSC1) gene, whose
cytogenetic location is 9p21.2, was first identified as a potential
lung cancer tumor suppressor gene in 2004 (25). Although TUSC1 was recently reported
to suppress cell proliferation and tumorigenicity in non-small cell
lung cancer, expression and epigenetic alteration of the TUSC1 gene
in gastroenterological cancer including HCC have not been
investigated (26,27). Accordingly, we focused on TUSC1 and
investigated the expression and regulatory mechanisms of TUSC1 in
order to determine if TUSC1 is a TSG that is silenced through
hypermethylation and if it is a novel epigenetic biomarker for
hepatocarcinogenesis and HCC progression.
Materials and methods
Sample collection
Nine HCC cell lines (Hep3B, HepG2, HLE, HLF, HuH1,
HuH2, HuH7, PLC/PRF/5 and SK-Hep1) were obtained from the American
Type Culture Collection (Manassas, VA, USA), stored at −80°C in a
cell preservative solution (=Tokyo, Japan) and cultured in
RPMI-1640 (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10%
fetal bovine serum at 37°C in a 5% CO2 atmosphere.
A 68-year-old woman with chronic hepatitis C was
discovered to have an HCC, 3 cm in diameter, on the right lobe of
her liver. A contrast-enhanced abdominal computed tomography (CT)
scan showed a homogeneous mass. The patient underwent partial
hepatectomy in 2007. Specimens from her tumor and corresponding
non-cancerous tissue were immediately harvested, and total RNA was
extracted for use in microarrays. The tumor was pathologically
diagnosed as HCC, and an area containing >80% of cancer cells
was selected for RNA extraction.
Primary HCC tissues and corresponding non-cancerous
tissues were collected consecutively from 94 patients undergoing
liver resection for HCC at Nagoya University Hospital between
January 1998 and July 2008. Specimens were classified
histologically according to the 7th edition of the Union for
International Cancer Control (UICC) classification (28). Background liver status, Child-Pugh
classification, hepatitis virus infection, preoperative serum tumor
markers, tumor multiplicity and maximum size, pathological findings
including tumor differentiation and vascular invasion were
investigated. The median duration of patient follow-up was 41.1
months (range, 0.8–147 months). Postoperative follow-up included
physical examinations and measurement of serum tumor markers every
three months, and enhanced CT scans (chest and abdominal cavity)
every six months. Treatment after recurrence was generally selected
from the following options: surgery, radiofrequency ablation,
transcatheter arterial chemoembolization and chemotherapy according
to tumor status and liver function.
Collected tissue samples were immediately flash
frozen in liquid nitrogen and stored at −80°C until RNA extraction
(28 days on average) was performed. Tumor samples, ~5 mm square,
without the necrotic component and confirmed to contain >80%
tumor cells by definition, were used for RNA extraction.
Corresponding non-cancerous liver tissue samples, collected >2
cm away from the edge of the tumor, were obtained from the same
patient and did not contain any regenerative or dysplastic
nodules.
The study fully conformed to the ethical guidelines
of the World Medical Association Declaration of Helsinki Ethical
Principles for Medical Research Involving Human Subjects. Written
informed consent for usage of clinical samples and data, as
required by the Institutional Review Board at Nagoya University,
Japan, was obtained from all enrolled patients.
Microarray procedure
Total RNA was isolated from each of the frozen
samples using the RNeasy mini kit (Qiagen, Chatsworth, CA, USA)
according to the manufacturer’s protocol. Gene expression profiles
were determined using Affymetrix HGU133A and HGU133B GeneChips
(Affymetrix, Santa Clara, CA, USA) according to the manufacturer’s
recommendations. In brief, double-stranded cDNA was synthesized
from 8 μg of total RNA with oligo T7-(dt)24 as the primer and
transcribed into biotinylated cRNA using an IVT labeling kit
(Affymetrix). Biotinylated cRNA (20 μg) was fragmented at 94°C for
35 min and hybridized to the human genome U133 Plus 2.0 gene chip
array (Affymetrix), which contains >54,000 probe sets. The cRNA
probes that hybridized to the oligonucleotide arrays were stained
with streptavidin R-phycoerythrin and processed for signal
intensity using Micro Array Suite 5.0 software (Affymetrix). All
data used for subsequent analysis passed the quality control
criteria.
Reverse transcription-polymerase chain
reaction (RT-PCR) and quantitative real-time RT-PCR
TUSC1 mRNA expression was analyzed using RT-PCR and
quantitative real-time RT-PCR. Total RNA (10 μg) was isolated from
HCC cell lines (Hep3B, HepG2, HLE, HLF, HuH1, HuH2, HuH7, PLC/PRF/5
and SK-Hep1), 94 primary HCCs and corresponding adjacent
non-cancerous tissues and was used to generate cDNAs. They were
then amplified using PCR primers for TUSC1; sense
(S)(5′-ACATGTACAGTTCCCCTGCC-3′ in exon 1) and antisense (AS)
(5′-GTGTTTCTTGGCACCCAGTT-3′ in exon 1), which amplify a 110 base
pair (bp) product. The RT-PCR amplification protocol was: 40 cycles
at 94°C for 15 sec, 60°C for 15 sec, and 72°C for 20 sec, after an
initial denaturation step at 94°C for 2 min. RT-PCR of β-actin was
also performed to confirm that equal amounts of cDNA were used as
templates. Each RT-PCR product was loaded directly onto 2% agarose
gels, stained with ethidium bromide and visualized under UV
illumination. Quantitative real-time RT-PCR was performed using the
SYBR-Green PCR core reagents kit (Perkin-Elmer, Applied Biosystems,
Foster City, CA, USA) under the following conditions: one cycle at
95°C for 10 min, then 45 cycles at 95°C for 15 sec and 60°C for 30
sec. Real-time detection of the SYBR-Green emission intensity was
conducted using an ABI prism 7000 Sequence Detector (Perkin-Elmer,
Applied Biosystems). The primers for the PCR reaction were the same
primers used for RT-PCR, as described above. For standardization
purposes, the expression of glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) mRNA (TaqMan, GAPDH control reagents, Applied
Biosystems) was quantified in each sample. Quantitative RT-PCR was
performed using nine HCC cell lines and 94 clinical samples in
triplicate and included samples without templates as negative
controls. The expression level of each sample is shown as the value
of TUSC1 divided by that of GAPDH.
Surveillance of the CpG island around the
TUSC1 gene locus
The base sequence around the TUSC1 gene was analyzed
for the presence of CpG islands to evaluate methylation as a
potential regulatory mechanism of TUSC1 gene expression. The
presence or absence of a CpG island was determined using the
following criteria: at least 200-bp region of DNA with a high GC
content (>50%) and an observed CpG/expected CpG ratio of ≥0.6
(29). We used the CpG Island
Searcher (http://cpgislands.usc.edu/) to
determine the location of putative CpG islands (30).
Methylation-specific PCR (MSP)
DNA samples extracted from HCC cell lines, clinical
HCC tissues and corresponding non-cancerous tissues were subjected
to bisulfite treatment. Briefly, 2 μg of DNA was denatured with
NaOH and modified using sodium bisulfite. DNA samples were then
purified using Wizard purification resin (Promega, Madison, WI,
USA), treated again with NaOH, precipitated with ethanol, and
resuspended in water. They were then amplified using unmethylated
PCR primers located in exon 1 of the TUSC1 gene: S
(5′-TGAGAGGATGAGTTGGGTAG-3′) and AS (5′-CCCCACTCAAACATAATCCC-3′),
which amplify a 121 bp product. Primers used to detect methylated
DNA were: S (5′-CGAGAGGACGAGTCGGGTAG-3′) and AS (5′-CGC
TCGAACGTAATCCCCGC-3′) both in exon 1, which amplify a 118 bp
product. The PCR amplification consisted of 40 cycles at 94°C for
15 sec, 60°C for 15 sec, and 72°C for 20 sec, following an initial
denaturation step at 94°C for 2 min. Each PCR product was loaded
directly onto 2% agarose gels, stained with ethidium bromide and
visualized under UV illumination.
Bisulfite sequence analysis
Genomic bisulfite-treated DNAs from HCC cell lines
were sequenced to verify the accuracy of the MSP results. The
primer pair used to generate a fragment for sequencing was in exon
1 of TUSC1: S (5′-GGTAGTTT TAGGGTTTTGAG-3′) and AS
(5′-AAACTACTCCTCC TTATCCC-3′), which amplifies a 121 bp product.
The PCR amplification protocol was as follows: 50 cycles at 94°C
for 20 sec, 60°C for 20 sec, and 72°C for 20 sec, following an
initial denaturation step at 94°C for 2 min. PCR products were
purified directly using the QIAquick PCR Purification kit (Qiagen,
Hilden, Germany). Purified DNA fragments were subcloned into the TA
cloning vector (Invitrogen, Carlsbad, CA, USA). Each cloned DNA was
mixed with 3 μl of specific primer (M13) and 4 μl of Cycle Sequence
Mix (ABI PRISM Terminator v1. 1 Cycle Sequencing kit; Applied
Biosystems). Sequence analysis was carried out using an Applied
Biosystems ABI310 and sequence electropherograms were generated
using ABI Sequence Analysis 3.0 software.
5-Aza-2′-deoxycytidine (5-aza-dC)
treatment
To assess the relationship between hypermethylation
and TUSC1 mRNA expression, HCC cell lines were treated with the DNA
methylation inhibitor 5-aza-dC (Sigma-Aldrich). Cells
(1.5×106) were cultured for 6 days with medium changes
on days 1, 3 and 5. After incubation, the cells were harvested, RNA
was extracted, and RT-PCR was performed as described above.
Immunohistochemical staining
We used immunohistochemical staining to investigate
TUSC1 protein localization in 35 representative HCC samples whose
sections were well preserved. Formalin-fixed, paraffin-embedded
tissue samples were dewaxed in xylene twice for 5 min, rehydrated
in graded alcohols 100, 90 and 70% and H2O for 2 min
each and subsequently treated with 3% H2O2 to
inhibit endogenous peroxidases, followed by antigen retrieval with
10 mM citrate buffer at 95°C for 5 min, repeated five times. The
samples were incubated with Histofine SAB-PO® (Nichirei,
Tokyo, Japan) for 5 min to limit nonspecific reactivity, and were
then incubated for 1 h at room temperature with a rabbit antibody
to TUSC1 (bs-6114R, Bioss Inc., Woburn, MA, USA), diluted 1:250 in
ChemMatet antibody diluent (Dako, Carpinteria, CA, USA). Samples
were then washed with phosphate-buffered saline, followed by 10 min
incubation with biotinylated secondary antibody (Histofine SAB-PO,
Nichirei). Sections were subsequently developed for 1 min using
liquid 3,3′-diaminobenzidine (DAB) as the substrate (Nichirei).
Staining properties were determined using blood vessels as internal
controls, and staining patterns in HCCs were compared with
corresponding non-cancerous tissues. To avoid subjectivity,
specimens were randomized and coded before analysis by two
independent observers, blinded to the status of the samples. Each
observer evaluated all specimens at least twice within a given time
interval to minimize intra-observer variation.
Statistical analysis
The relative mRNA expression levels (TUSC1/GAPDH)
were calculated from quantified data. Differences in TUSC1
expression levels between two groups were analyzed using the
Mann-Whitney U test. The association between the methylation status
of TUSC1 and clinicopathological parameters was evaluated using the
χ2 test. Disease-specific survival rates were calculated
using the Kaplan-Meier method, and the difference in survival
curves was analyzed using the generalized Wilcoxon test. All
statistical analysis was performed using JMP® 10
software (SAS Institute Inc., Cary, NC, USA). A p-value of <0.05
was considered to indicate statistically significant
differences.
Results
Patient characteristics
The ages of the 94 patients ranged from 34–84 years
(64.5±10.0 years, mean ± SD), and the male to female ratio was
77:17. Twenty-six patients had hepatitis B and 53 had hepatitis C
infections. In terms of the non-cancerous liver, the number of
patients with normal liver, chronic hepatitis and cirrhosis were 9,
49 and 36, respectively. Eighty-seven patients were in Child-Pugh
class A and 12 patients were in class B. When classified according
to the 7th edition of the UICC classification, 11, 44, 29 and 10
patients were in stages I, II, III and IV, respectively.
Expression array
We conducted an expression array in order to find a
new candidate TSG in HCC. We searched for genes whose expression in
tumor tissues was reduced further than that in corresponding
non-cancerous tissues, and TUSC1 expression was found to be reduced
in HCC compared with normal tissue with a log2 ratio of -3.4
(Table I).
 | Table IMicroarray results for TUSC1
expression. |
Table I
Microarray results for TUSC1
expression.
| Gene symbol | Log2 ratio | Normal signal | Detection | Tumor signal | Detection | Probe ID | Chromosomal
location |
|---|
| TUSC1 | −3.4 | 252.1 | P | 25.6 | P | HU133p2_36644 | Chr 9p21.2 |
TUSC1 mRNA expression analyzed by
quantitative RT-PCR
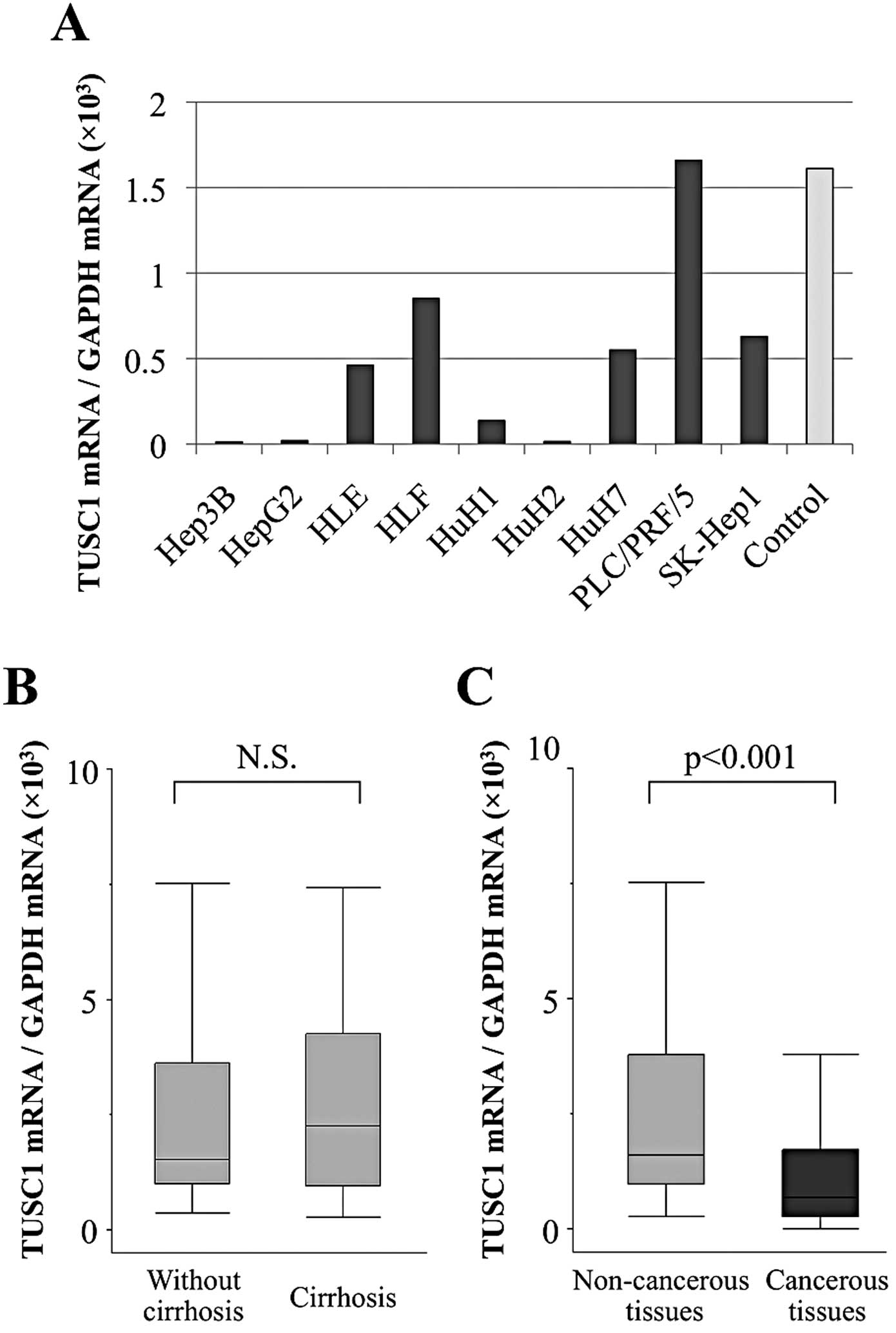
Reduced expression of TUSC1 mRNA was confirmed in
all HCC cell lines, except for PLC/PRF/5, when compared with the
median expression level in non-cancerous liver tissues (Fig. 1A). In particular, Hep3B, HepG2, HuH1
and HuH2 showed marked suppression of TUSC1 mRNA.
When TUSC1 mRNA expression levels in non-cancerous
tissue samples without cirrhosis (n=58) were compared to those with
cirrhosis (n=36), no significant differences were found, suggesting
that the expression of TUSC1 mRNA in non-cancerous liver was not
affected by background liver fibrosis (Fig. 1B). In 79 (84%) of 94 patients, the
TUSC1 mRNA expression level was lower in HCC tissues than in
corresponding normal tissues. Additionally, mean expression of
TUSC1 mRNA was significantly lower in HCC tissues than in
corresponding normal tissues (p<0.001; Fig. 1C).
Identification of a CpG island
TUSC1 is an intronless gene. The CpG island was
found inside exon 1 of the TUSC1 gene using the CpG Island
Searcher; length, 1,315 bp; GC content 67.8% and an observed
CpG/expected CpG ratio of 0.82 (Fig.
2). Accordingly, we hypothesized that hypermethylation of the
intragenic CpG island is the mechanism responsible for regulating
TUSC1 gene expression in HCC tissue.
MSP of HCC cell lines
MSP was conducted to verify the above hypothesis. We
first examined the methylation status of nine HCC cell lines.
Appropriate-sized MSP bands were confirmed in Hep3B, HepG2 and
HuH2. Following PCR using unmethylated primers, appropriate-sized
bands were confirmed in all HCC cell lines other than Hep3B and
HuH2 (Fig. 3A). We concluded that
complete methylation occurred in Hep3B and HuH2, with an absence of
methylation in HLE, HLF, HuH1, HuH7, PLC/PRF/5 and SK-Hep1, and
only partial methylation in HepG2.
Expression of TUSC1 after 5-aza-dC
treatment
To explore whether intragenic hypermethylation leads
to the suppression of expression, we examined the expression of
TUSC1 in HCC cell lines before and after treatment with the DNA
methylation inhibitor, 5-aza-dC. Applying semi-quantitative RT-PCR,
a reactivation or increase in TUSC1 expression was shown to occur
in Hep3B, HepG2 and HuH2 cells in agreement with the MSP results
(Fig. 3B).
Bisulfite sequence analysis
To ascertain whether the MSP amplification was
performed reliably, we conducted a direct sequence analysis of HuH2
(complete methylation in MSP) and HLF (absence of methylation in
MSP). We found that all CpGs in the HuH2 fragment were CG, while
all those in HLF were TG (Fig. 3C).
These results indicated that the MSP had worked correctly.
Expression and methylation status of
TUSC1 in 94 clinical HCC samples
Downregulation of TUSC1 mRNA was observed in 79 of
94 (84.0%) tumor samples from patients with HCC and there was no
significant association with overall or recurrence-free
survival.
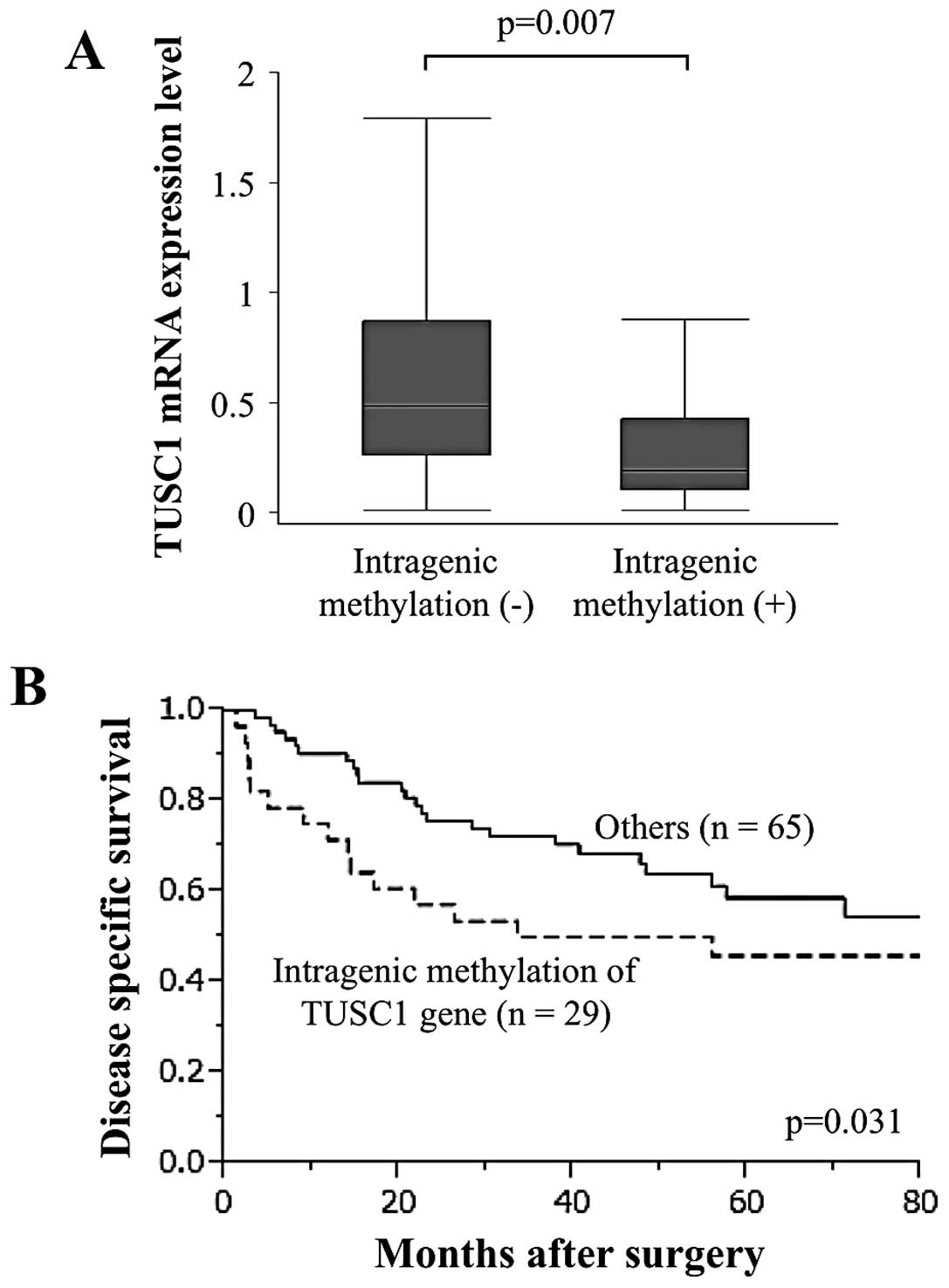
Using MSP, 29 (30.9%) of 94 HCC tissues and only two
(2.1%) of 94 corresponding non-cancerous tissues showed intragenic
hypermethylation in the TUSC1 gene. The TUSC1 mRNA expression level
in HCC patients with intragenic methylated TUSC1 was significantly
lower than in those without, indicating that intragenic methylation
contributed to the strong suppression of TUSC1 transcription
(Fig. 4A). Moreover,
disease-specific survival was significantly shorter in patients
with intragenic hypermethylation of TUSC1 than in those without
(p=0.031; Fig. 4B).
Analysis of the association between the methylation
status of TUSC1 and clinicopathological factors including
demographics, background liver status and pathological findings
showed that intragenic hypermethylation of TUSC1 in HCC was
significantly associated with advanced UICC stage (p=0.025;
Table II).
| Table IIAssociation between methylation
status of TUSC1 and clinicopathological parameters in 94 HCC
patients. |
Table II
Association between methylation
status of TUSC1 and clinicopathological parameters in 94 HCC
patients.
| Clinicopathological
parameters | Methylation
positive in tumor tissues (n) | Methylation
negative in tumor tissues (n) | p-value |
|---|
| Age |
| <65 year | 16 | 31 | 0.503 |
| ≥65 year | 13 | 34 | |
| Gender |
| Male | 22 | 55 | 0.318 |
| Female | 7 | 10 | |
| Background
liver |
| Normal liver | 2 | 7 | 0.204 |
| Chronic
hepatitis | 12 | 37 | |
| Cirrhosis | 15 | 21 | |
| Child-Pugh
classification |
| A | 27 | 60 | 0.891 |
| B | 2 | 5 | |
| Hepatitis
virus |
| Absent | 2 | 13 | 0.199 |
| HBV | 10 | 16 | |
| HCV | 17 | 36 | |
| AFP (ng/ml) |
| ≤20 | 12 | 35 | 0.263 |
| >20 | 17 | 30 | |
| PIVKA II
(mAU/ml) |
| ≤40 | 11 | 29 | 0.544 |
| >40 | 18 | 36 | |
| Tumor
multiplicity |
| Solitary | 20 | 50 | 0.419 |
| Multiple | 9 | 15 | |
| Tumor size |
| <3.0 cm | 7 | 20 | 0.507 |
| ≥3.0 cm | 22 | 45 | |
|
Differentiation |
| Well | 6 | 21 | 0.241 |
| Moderate to
poor | 23 | 44 | |
| Growth type |
| Expansive
growth | 23 | 54 | 0.664 |
| Invasive
growth | 6 | 11 | |
| Serosal
infiltration |
| Absent | 20 | 49 | 0.519 |
| Present | 9 | 16 | |
| Formation of
capsule |
| Absent | 8 | 18 | 0.992 |
| Present | 21 | 47 | |
| Infiltration to
capsule |
| Absent | 13 | 27 | 0.766 |
| Present | 16 | 38 | |
| Septum
formation |
| Absent | 10 | 24 | 0.820 |
| Present | 19 | 41 | |
| Vascular
invasion |
| Absent | 22 | 49 | 0.960 |
| Present | 7 | 16 | |
| UICC pathological
stage |
| I, II | 12 | 43 | 0.025a |
| III, IV | 17 | 22 | |
Immunohistochemical staining
Next, immunohistochemical staining was performed to
examine the expression of the TUSC1 protein in cases showing
underexpressed and equivalent TUSC1 mRNA levels in HCC tissues
relative to corresponding non-cancerous tissues. Two representative
cases with downregulation of TUSC1 mRNA expression in HCC tissues
showed reduced expression of TUSC1 protein in the cytoplasm of
cancerous components compared with adjacent non-cancerous tissue
components (Fig. 5A and B).
Equivalent expression of TUSC1 protein both in cancerous and
non-cancerous components was confirmed in the case without
downregulation of TUSC1 mRNA expression in HCC (Fig. 5C). MSP results of the cases
presented are shown in Fig. 5D.
Overall, results of immunohistochemical staining were consistent
with quantitative real-time PCR data for TUSC1.
Discussion
In the present study, we investigated the expression
and methylation status of TUSC1 identified as a candidate TSG in
HCC based on our microarray results. Consequently, the attenuated
mRNA expression of TUSC1 in most of the cancer tissues compared
with non-cancerous adjacent tissues implicated its role as a TSG.
Given that TUSC1 mRNA expression was independent of progression of
hepatic fibrosis, suppression of TUSC1 could be considered as a
specific event that occurs in the final stage in the development of
HCC.
Our analysis of the TUSC1 gene sequence showing that
the gene possessed a CpG island inside exon 1 suggested that
aberrant methylation was a potential mechanism regulating TUSC1
gene expression, although there have been no reports related to
methylation analysis of the TUSC1 gene. TUSC1 transcription in
Hep3B, HepG2 and HuH2 cells was markedly reduced, coinciding with
intragenic hypermethylation. In surgical specimens, HCC tissues
with intragenic hypermethylation of the TUSC1 gene displayed
significantly lower expression of TUSC1 mRNA, and were associated
with advanced UICC stage and, subsequently, an adverse prognosis.
These results indicated that intragenic hypermethylation of the
TUSC1 gene downregulated transcription and could be useful as a
prognostic marker of HCC.
Alteration of chromosome 9p is implicated in a
variety of tumor types including HCC through chromosomal
inversions, translocations, loss of heterozygosity (LOH) and
homozygous deletion, suggesting that chromosome 9p contains a tumor
suppressor locus critical in the development of tumors (31–34).
Two candidate tumor suppressor loci were identified in the
chromosome 9p21 region. One locus was p16/CDKN2A and the other
p15/CDKN2B (35,36). P16/CDKN2A is one of the major TSGs
(32,34,35).
The TUSC1 gene was newly identified on chromosome 9p21.2 in 2004
and found to have tumor suppressor activity in lung cancer
(25,27). The TUSC1 locus is distinct from that
of p16/CDKN2A and approximately 3.7 Mb proximal to p16/CDKN2A
(25). TUSC1 has unique features
including an intronless structure and an intragenic CpG island as
shown in Fig. 2. Although potential
roles played by the TUSC1 gene have been investigated only in lung
cancer, our results provide new insight into understanding the
function of TUSC1.
As shown in this study, TUSC1 possesses a CpG island
inside exon 1, not in the promoter region, and intragenic
hypermethylation was related to downregulation of TUSC1 mRNA.
Recently, the role of intragenic methylation has led to a heated
discussion amongst researchers. Since the 1970s, DNA methylation
has been described as a silencing epigenetic mark that occurred in
the promoter region (37).
Improvement of genome-scale mapping of methylation has enabled the
evaluation of DNA methylation in the following different genomic
contexts: transcriptional start sites with or without CpG islands,
in gene bodies, in regulatory elements and in repeat sequences
(37,38). In recent studies, the majority of
methylated CpG islands were shown to be in intragenic and
intergenic regions, whereas <3% of CpG islands in 5′ promoters
were methylated (38).
Although it is known that the methylation of DNA in
promoter regions suppresses gene transcription, the role of DNA
methylation in gene bodies remains unclear. Tissue-specific
intragenic methylation might reduce, or, paradoxically, enhance
transcription elongation efficiency (39–41).
In this study, intragenic hypermethylation of the TUSC1 gene was
confirmed in some HCC cell lines and clinical HCC tissues and was
related to downregulation of TUSC1 gene transcription. However, it
is highly controversial if hypermethylation of the TUSC1 gene can
be considered as intragenic methylation, as is the case with other
genes that possess introns and are of longer gene lengths, as the
TUSC1 gene is intronless and consists of only 2,461 bases. There is
a possibility that the transcription regulatory site and
transcription site are overlapping one another. Although our
results demonstrated that methylation of the TUSC1 gene played a
role in transcriptional regulation, intragenic hypermethylation was
not always confirmed in HCC cell lines and clinical HCC tissues in
which downregulation of TUSC1 mRNA occurred. LOH could be another
regulatory factor of TUSC1 transcription since chromosome 9p was
reported to be a frequent site of LOH in HCC (33).
This study is limited by its lack of sufficient
functional analysis of the TUSC1 gene; thus, we were unable to
state conclusively that TUSC1 is a TSG in HCC. Further studies are
required to clarify the molecular mechanisms underlying the
biological activities of TUSC1 in HCC.
In summary, TUSC1 is a putative TSG in HCC.
Intragenic methylation of the TUSC1 gene was one of the regulatory
mechanisms of TUSC1 mRNA transcription and TUSC1 could be a novel
prognostic marker of HCC.
References
|
1
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar
|
|
2
|
Hernandez-Gea V, Toffanin S, Friedman SL
and Llovet JM: Role of the microenvironment in the pathogenesis and
treatment of hepatocellular carcinoma. Gastroenterology.
144:512–527. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
4
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang JD and Roberts LR: Hepatocellular
carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 7:448–458.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kanda M, Nomoto S, Nishikawa Y, Sugimoto
H, Kanazumi N, Takeda S and Nakao A: Correlations of the expression
of vascular endothelial growth factor B and its isoforms in
hepatocellular carcinoma with clinico-pathological parameters. J
Surg Oncol. 98:190–196. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sanyal AJ, Yoon SK and Lencioni R: The
etiology of hepatocellular carcinoma and consequences for
treatment. Oncologist. 15:14–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Khare S, Zhang Q and Ibdah JA: Epigenetics
of hepatocellular carcinoma: Role of microRNA. World J
Gastroenterol. 19:5439–5445. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Villanueva A, Hoshida Y, Toffanin S,
Lachenmayer A, Alsinet C, Savic R, Cornella H and Llovet JM: New
strategies in hepatocellular carcinoma: genomic prognostic markers.
Clin Cancer Res. 16:4688–4694. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seitz HK and Stickel F: Risk factors and
mechanisms of hepatocarcinogenesis with special emphasis on alcohol
and oxidative stress. Biol Chem. 387:349–360. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levental KR, Yu H, Kass L, Lakins JN,
Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W,
Yamauchi M, Gasser DL and Weaver VM: Matrix crosslinking forces
tumor progression by enhancing integrin signalling. Cell.
139:891–906. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yates LR and Campbell PJ: Evolution of the
cancer genome. Nat Rev Genet. 13:795–806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Coulon S, Heindryckx F, Geerts A, Van
Steenkiste C, Colle I and Van Vlierberghe H: Angiogenesis in
chronic liver disease and its complications. Liver Int. 31:146–162.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sawan C, Vaissiere T, Murr R and Herceg Z:
Epigenetic drivers and genetic passengers on the road to cancer.
Mutat Res. 642:1–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanda M, Nomoto S, Okamura Y, Hayashi M,
Hishida M, Fujii T, Nishikawa Y, Sugimoto H, Takeda S and Nakao A:
Promoter hypermethylation of fibulin 1 gene is associated with
tumor progression in hepatocellular carcinoma. Mol Carcinog.
50:571–579. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kanda M, Nomoto S, Okamura Y, Nishikawa Y,
Sugimoto H, Kanazumi N, Takeda S and Nakao A: Detection of
metallothionein 1G as a methylated tumor suppressor gene in human
hepatocellular carcinoma using a novel method of double combination
array analysis. Int J Oncol. 35:477–483. 2009.
|
|
18
|
Dango S, Mosammaparast N, Sowa ME, Xiong
LJ, Wu F, Park K, Rubin M, Gygi S, Harper JW and Shi Y: DNA
unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA
alkylation repair and cancer cell proliferation. Mol Cell.
44:373–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Youngblood B, Oestreich KJ, Ha SJ,
Duraiswamy J, Akondy RS, West EE, et al: Chronic virus infection
enforces demethylation of the locus that encodes PD-1 in
antigen-specific CD8(+) T cells. Immunity. 35:400–412.
2011.PubMed/NCBI
|
|
20
|
Um TH, Kim H, Oh BK, Kim MS, Kim KS, Jung
G and Park YN: Aberrant CpG island hypermethylation in dysplastic
nodules and early HCC of hepatitis B virus-related human multistep
hepatocarcinogenesis. J Hepatol. 54:939–947. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lao V and Grady W: Epigenetics and
colorectal cancer. Nat Rev Gastroenterol Hepatol. 8:686–700. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yuan Y, Wang J, Li J, Wang L, Li M, Yang
Z, Zhang C and Dai JL: Frequent epigenetic inactivation of spleen
tyrosine kinase gene in human hepatocellular carcinoma. Clin Cancer
Res. 12:6687–6695. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moribe T, Iizuka N, Miura T, Kimura N,
Tamatsukuri S, Ishitsuka H, Hamamoto Y, Sakamoto K, Tamesa T and
Oka M: Methylation of multiple genes as molecular markers for
diagnosis of a small, well-differentiated hepatocellular carcinoma.
Int J Cancer. 125:388–397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kanda M, Nomoto S, Oya H, Takami H, Hibino
S, Hishida M, Suenaga M, Yamada S, Inokawa Y, Nishikawa Y, Asai M,
Fujii T, Sugimoto H and Kodera Y: Downregulation of DENND2D
by promoter hypermethylation is associated with early recurrence of
hepatocellular carcinoma. Int J Oncol. 44:44–52. 2014.
|
|
25
|
Shan Z, Parker T and Wiest JS: Identifying
novel homozygous deletions by microsatellite analysis and
characterization of tumor suppressor candidate 1 gene, TUSC1, on
chromosome 9p in human lung cancer. Oncogene. 23:6612–6620. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang XR, Liang X, Pfeiffer RM, Wheeler W,
Maeder D, Burdette L, Yeager M, Chanock S, Tucker MA and Goldstein
AM: Associations of 9p21 variants with cutaneous malignant
melanoma, nevi, and pigmentation phenotypes in melanoma-prone
families with and without CDKN2A mutations. Fam Cancer. 9:625–633.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shan Z, Shakoori A, Bodaghi S, Goldsmith
P, Jin J and Wiest JS: TUSC1, a putative tumor suppressor gene,
reduces tumor cell growth in vitro and tumor growth in vivo. PLoS
One. 8:e661142013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sobin LH, Gospodarowicz MK and Wittekind
C: UICC TNM classification of malignant tumours. 7th edition.
Wiley-Liss; New York: 2009
|
|
29
|
Takai D and Jones PA: Comprehensive
analysis of CpG islands in human chromosomes 21 and 22. Proc Natl
Acad Sci USA. 99:3740–3745. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takai D and Jones PA: The CpG island
searcher: a new WWW resource. In Silico Biol. 3:235–240.
2003.PubMed/NCBI
|
|
31
|
Mead LJ, Gillespie MT, Hung JY, Rane US,
Rayeroux KC, Irving LB and Campbell LJ: Frequent loss of
heterozygosity in early non-small cell lung cancers at chromosome
9p21 proximal to the CDKN2a gene. Int J Cancer. 71:213–217. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wiest JS, Franklin WA, Otstot JT, Forbey
K, Varella-Garcia M, Rao K, Drabkin H, Gemmill R, Ahrent S,
Sidransky D, Saccomanno G, Fountain JW and Anderson MW:
Identification of a novel region of homozygous deletion on
chromosome 9p in squamous cell carcinoma of the lung: the location
of a putative tumor suppressor gene. Cancer Res. 57:1–6.
1997.PubMed/NCBI
|
|
33
|
Sheu JC, Lin YW, Chou HC, Huang GT, Lee
HS, Lin YH, Huang SY, Chen CH, Wang JT, Lee PH, Lin JT, Lu FJ and
Chen DS: Loss of heterozygosity and microsatellite instability in
hepatocellular carcinoma in Taiwan. Br J Cancer. 80:468–476. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pollock P, Welch J and Hayward N: Evidence
for three tumor suppressor loci on chromosome 9p involved in
melanoma development. Cancer Res. 61:1154–1161. 2001.PubMed/NCBI
|
|
35
|
Cairns P, Polascik TJ, Eby Y, Tokino K,
Califano J, Merlo A, Mao L, Herath J, Jenkins R, Westra W, Rutter
J, Buckler A, Gabrielson E, Tockman M, Cho K, Hedrick L, Bova G,
Isaacs W, Koch W, Schwab D and Sidransky D: Frequency of homozygous
deletion at p16/CDKN2 in primary human tumours. Nat Genet.
11:210–212. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Serrano M, Hannon GJ and Beach D: A new
regulatory motif in cell-cycle control causing specific inhibition
of cyclin D/CDK4. Nature. 366:704–707. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jones PA: Functions of DNA methylation:
islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maunakea AK, Nagarajan RP, Bilenky M,
Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C,
Zhao Y, Turecki G, Delaney A, Varhol R, Thiessen N, Shchors K,
Heine VM, Rowitch DH, Xing X, Fiore C, Schillebeeckx M, Jones SJ,
Haussler D, Marra MA, Hirst M, Wang T and Costello JF: Conserved
role of intragenic DNA methylation in regulating alternative
promoters. Nature. 466:253–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Flanagan J and Wild L: An epigenetic role
for noncoding RNAs and intragenic DNA methylation. Genome Biol.
8:3072007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lorincz M, Dickerson D, Schmitt M and
Groudine M: Intragenic DNA methylation alters chromatin structure
and elongation efficiency in mammalian cells. Nat Struct Mol Biol.
11:1068–1075. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ball M, Li JB, Gao Y, Lee JH, LeProust EM,
Park IH, Xie B, Daley GQ and Church GM: Targeted and genome-scale
strategies reveal gene-body methylation signatures in human cells.
Nat Biotechnol. 27:361–368. 2009. View Article : Google Scholar : PubMed/NCBI
|