Introduction
Salivary gland cancer (SGC) is a rare cancer type of
the head and neck region where cancer cells form in major or minor
salivary glands (1–3). Generally, surgical resection is
considered to be the most common and successful therapeutic
approach for SGC performed today (1,4);
however, SGC frequently recurs locally and metastasises to the
regional lymph nodes and distant organs, which results in poor
prognosis (4–6). Although other treatment approaches for
SGC may include radiation therapy and chemotherapy, SGC exhibits
low sensitivity to irradiation and chemotherapy. Therefore, the
development of more effective chemotherapeutic drugs and strategies
against SGC are desirable.
Cancer has long been known to be a primarily genetic
disease initiated and progressed by alterations in genes, such as
oncogenes and tumour suppressor genes. However, recent reports
indicate that various types of cancers are caused by epigenetic
changes, which are heritable changes in gene expression that occur
without alteration in DNA sequences (7–9). These
epigenetic changes, including DNA methylation and histone
modifications, modify the structure of chromatin and affect the
accessibility of regulatory proteins to DNA (10,11).
Histone acetylation is one such epigenetic change and has been
reported to play important roles in the initiation and progression
of cancers (12,13). Histone deacetylation can cause the
tightening of chromatin and can block transcriptional activation.
Moreover, it is reversible and is regulated by a balance between
the opposing activities of histone acetyltransferase (HAT) and
histone deacetylase (HDAC). HAT is an enzyme that catalyses the
acetylation of N-terminal lysine residues in histone proteins,
which reduces the affinity of histone for DNA and increases the
accessibility of transcriptional regulatory protein to chromatin
(10). On the other hand, HDAC is
an enzyme that catalyses the removal of acetyl groups from
histones, and increased HDAC activities are usually associated with
transcriptional repression (10).
Recently, altered HDAC activities have been shown to
be present in many types of cancers, possibly representing an
attractive target for cancer therapy (7–9,12,13).
HDAC inhibitors induce cell cycle arrest and apoptosis of cancer
cells by blocking deacetylases leading to the accumulation of
hyperacetylated histones and alteration of gene transcription
(7,9,11,14,15,17,18).
Various classes of HDAC inhibitors have been identified and are
currently being tested in phase I/II clinical trials (12,13).
Valproic acid (VPA), which is widely used for epilepsy and
manic-depressive psychosis, has also recently garnered attention as
a specific inhibitor of HDAC activity (7,19). It
is known that chromatin with hyperacetylated core histones adopts a
relaxed conformation with the unfolding of the associated DNA and
chromatin remodelling during the nucleosome packing of DNA, which
are key steps in the regulation of many genes that play crucial
roles in cell growth and differentiation (10). VPA has been reported to show
antitumour effects against many types of cancers such as acute
myeloid leukaemia, breast and prostate cancers and head and neck
squamous cell carcinomas. The objective of the present study was to
define the biological and therapeutic effects of VPA in treating
SGC.
Materials and methods
Valproic acid
Valproic acid sodium salt (VPA; Sigma Aldrich Co.,
St. Louis, MO, USA) was dissolved in Dulbecco’s phosphate-buffered
saline (PBS) without Ca2+ or Mg2+ (DPBS;
Sigma Aldrich) as a stock solution. For in vitro
experiments, the VPA stock solution was diluted with the complete
culture medium. For in vivo experiments, the VPA stock
solution was supplemented in drinking water.
Cell lines and cell culture
Two human SGC cell lines, HSY and HSG cells, were
used in the present study. They are adenocarcinoma cell lines
derived from the parotid gland and submandibular gland,
respectively (20,21). These two cell lines produce tumours
when subcutaneously inoculated into nude mice. They were maintained
in Dulbecco’s modified Eagle’s medium (DMEM; Sigma Aldrich)
supplemented with 10% fetal calf serum (FCS), 100 μg/ml
streptomycin and 100 U/ml penicillin in a humidified atmosphere
composed of 95% air and 5% CO2 at 37°C.
In vitro cell proliferation assay
The proliferation of SGC cells treated with VPA was
assessed by MTT assay. Cells were seeded on 96-well plates (Falcon,
Franklin Lakes, NJ, USA) at 2×103 cells/well in DMEM
containing 10% FCS. Twenty-four hours later, cells were treated
with various concentrations (0, 1, 2, 5 and 10 mM) of VPA for 2 and
4 days. After VPA treatment, a 10 μl aliquot of 5 mg/ml
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma Aldrich) was added to each well, and the cells were
incubated for 4 h. The blue dye taken up by cells was dissolved in
dimethyl sulphoxide, and the absorbance was measured with a
microplate reader (Bio-Rad Laboratories, Hercules, CA, USA) at 540
nm. All assays were run in triplicate.
Mice and in vivo study
BALB/c nude mice were purchased from Japan CLEA Inc.
(Osaka, Japan). The mice were maintained under pathogen-free
conditions and were handled in accordance with the Guidelines for
Animal Experimentation of Tokushima University. The experiments
were initiated when the mice were 8 weeks of age and were performed
as previously described (23). Five
million HSY cells were inoculated subcutaneously into the backs of
mice through 26G needles. Once palpable tumours were established at
1 week after the inoculation, VPA treatment was started by
administering VPA (0, 0.2 and 0.4% w/v) in drinking water. The
tumour volume and body weight of the mice were measured twice a
week. Tumour volumes were calculated using the following formula:
Tumour volume = 0.5 × L × W2, where L and W represent
the largest diameter and the smallest diameter, respectively. For
each treatment, six mice were used.
Cell cycle analysis
The cell cycle distribution was analysed using a
fluorescence-activated cell sorter (FACS) as previously described
(23,24). Cells grown under subconfluent
conditions were treated with VPA (0, 2 and 5 mM). After 12 or 24 h
of VPA treatment, cells were collected and fixed with ice-cold 70%
ethanol for 30 min and stored at 4°C until further analysis. Then,
cells were treated with propidium iodide (40 μg/ml) and RNase A (1
μg/ml) for 20 min at 37°C. Samples were kept on ice, and the cell
cycle analysis was completed by measuring the propidium
iodide-stained DNA content with an Epics® XL-MCL
cytometer (Beckman Coulter, Brea, CA, USA).
Real-time reverse
transcription-polymerase chain reaction (RT-PCR)
Cancer cells grown under subconfluent conditions
were treated with VPA (0, 2 and 5 mM). After 24 or 48 h of VPA
treatment, total RNA was prepared from the cells using
Isogen® (Nippon Gene, Toyama, Japan) according to the
manufacturer’s instructions. The mRNA expression of p21 and p27 was
examined by real-time reverse transcriptase-polymerase chain
reaction (RT-PCR). Glyceraldehyde 3-phosphate dehydrogenase
(GAPDH), a housekeeping gene, was used as an internal control.
Gene-specific products were measured continuously by an ABI PRISM
7000 Sequence Detection System (Applied Biosystems) over 40 cycles.
Experiments were performed at least three times.
Statistical analysis
All numerical data are expressed as the average of
the values obtained ± SD. Significant differences between the means
for the different groups were evaluated using one-way ANOVA, with
the level of significance at P<0.05. All experiments were
repeated two to three times, and similar results were obtained.
Results
Effect of VPA on the proliferation of
salivary gland cancer cells in vitro
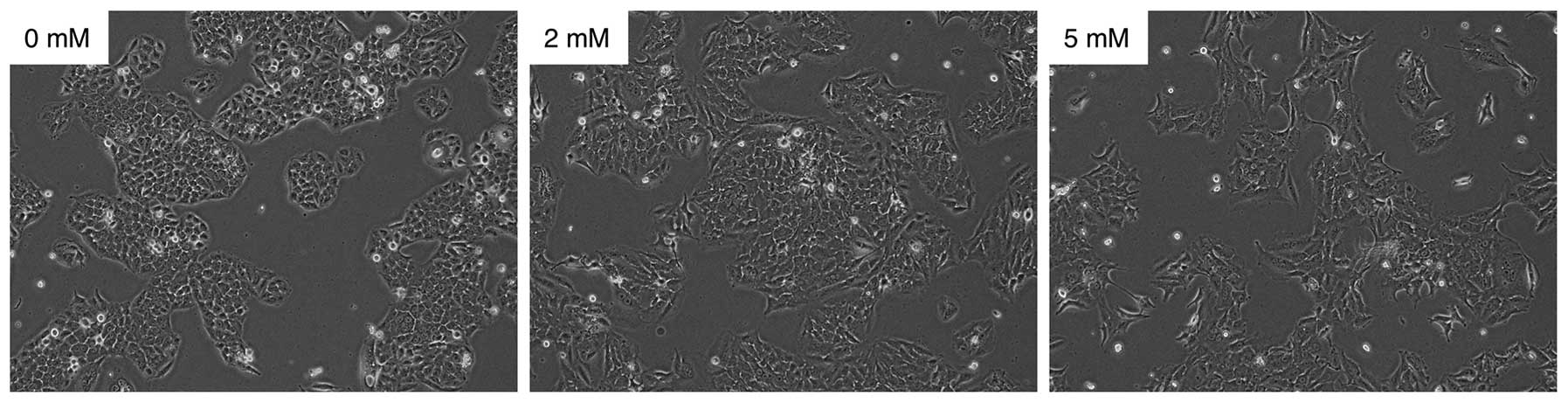
We examined the effect of VPA on cell morphology and
proliferation of SGC cells in vitro. The morphology of
cancer cells treated with 0, 2 and 5 mM of VPA for 24 h was
evaluated. As shown in Fig. 1, the
morphology of the HSY cells was transformed from being cuboidal to
being spindle-shaped in a dose-dependent manner. The results
obtained for the HSG cells were similar to those obtained for the
HSY cells (data not shown).
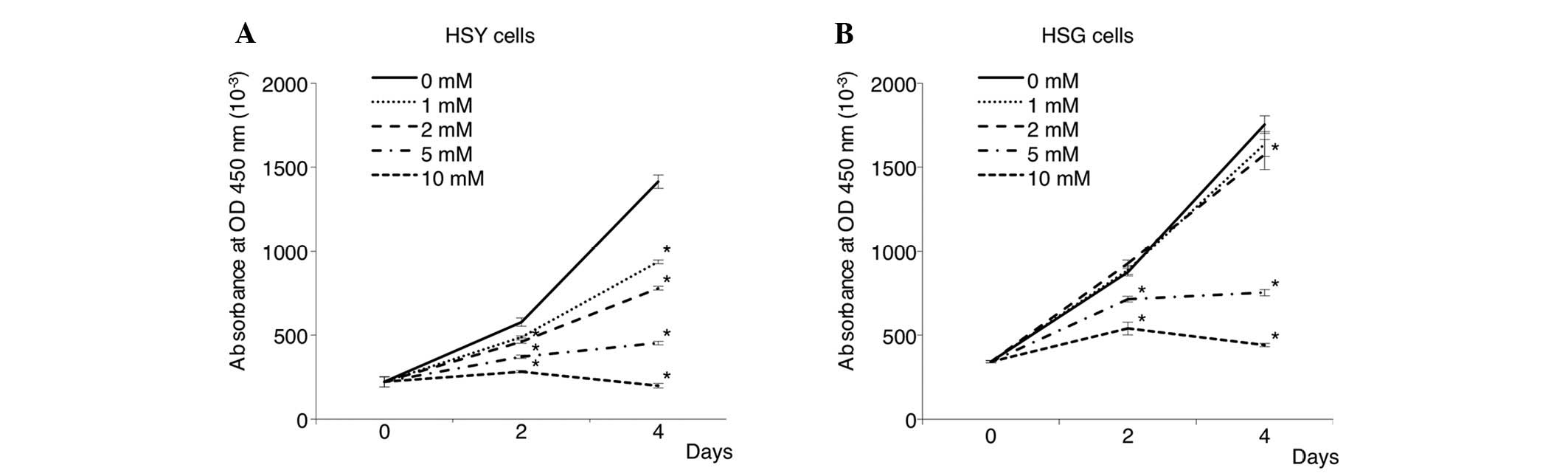
Next, the proliferation of cancer cells treated with
0, 1, 2, 5 and 10 mM of VPA was assessed by MTT assay. As shown in
Fig. 2, VPA inhibited the
proliferation of SGC cells in a dose-dependent manner. Treatment
with 5 mM VPA induced a proliferation inhibition rate of ~40–70% in
both cancer cell lines. Degenerated cancer cells were observed at a
high concentration (10 mM) of VPA. In particular, the HSY cells
were most sensitive to the inhibitory effects of VPA on cell
proliferation.
Antitumour effect of VPA on salivary
gland tumours in vivo
Based on the in vitro findings discussed
above, to investigate the antitumour effect of VPA on salivary
gland tumours in vivo, experiments with salivary gland
tumour xenografts were performed. HSY cells were inoculated
subcutaneously into the back of nude mice (n=5), and oral VPA (0.2
and 0.4% w/v) administration in drinking water was initiated at 1
week after the inoculation. Control mice drank water without VPA.
As shown in Fig. 3, VPA treatment
significantly suppressed tumour growth compared with the growth
observed in the untreated controls. However, there was no
significant difference between the 0.2 and 0.4% VPA groups. In
addition, all of the VPA-treated mice without inoculation of cancer
cells showed weight loss due to drug toxicity (data not shown).
Effect of VPA on the cell cycle
distribution of salivary gland cancer cells
We next investigated the effect of VPA on the cell
cycle distribution of SGC cells to explore the underlying mechanism
of VPA-mediated cell growth inhibition. Cancer cells treated with 2
and 5 mM of VPA were subjected to cell cycle analysis by flow
cytometry. As shown in Fig. 4, VPA
treatment effectively altered the cell cycle distribution of the
HSY cells. Compared to the controls showing 56.2% cells in the G1
phase at 24 h, VPA treatment at doses of 2 and 5 mM resulted in
60.4 (P<0.05) and 73.8% (P<0.05) cells in the G1 phase of the
cell cycle, respectively (Table I).
The population of cells in the G1 phase was significantly increased
in the VPA treatment group relative to the population in the
untreated controls. Similar results were also observed for HSG
cells (data not shown).
 | Table IEffect of VPA on cell cycle
distribution of salivary gland cancer cells (HSY cells). |
Table I
Effect of VPA on cell cycle
distribution of salivary gland cancer cells (HSY cells).
| | VPA |
|---|
| |
|
|---|
| | 2 mM | 5 mM |
|---|
| |
|
|
|---|
| Control (%) | 12 h (%) | 24 h (%) | 12 h (%) | 24 h (%) |
|---|
| G1 | 56.2±0.8 | 57.7±6.0 | 60.4±0.6a | 67.1±1.6a | 73.8±2.0a |
| G2 | 24.6±0.3 | 21.9±0.6 | 18.3±2.2a | 21.2±0.1a | 15.1±0.6a |
Mechanisms of the antitumour effect of
VPA on salivary gland cancers
To further identify the mechanisms of the antitumour
effect on SGC by VPA treatment, the expression levels of
tumour-suppressor genes p21 and p27 were examined by real-time
RT-PCR. Fig. 5 shows the mRNA
expression levels of p21 and p27 in HSY and HSG cells after 24 or
48 h of treatment with 2 or 5 mM VPA. VPA markedly upregulated the
levels of both p21 and p27 mRNA expression in both SGC cell lines.
The expression of p21 and p27 mRNA was increased in a
time-dependent manner. In particular, in HSY cells treated with 5
mM VPA, the mRNA expression levels of both p21 and p27 were
increased appreciably.
Discussion
SGC frequently spreads to regional lymph nodes and
distant organs and its prognosis is quite poor (1–6). Among
the multiple options available for SGC treatment, surgical
resection is currently the primary choice and standard option
(5,6). In addition, SGC shows low sensitivity
to chemotherapy and radiotherapy, and the effectiveness of systemic
chemotherapy against SGC has not been established. Therefore, it is
apparent that the development of more effective chemotherapeutic
drugs and strategies against SGC is necessary. Recent advancements
in the field of cancer epigenetics have shown that the development
of cancer involves epigenetic abnormalities along with genetic
alterations (9,10). Histone acetylation is one of these
epigenetic changes and a regulator of gene expression; moreover, it
plays an important role in carcinogenesis. Histone acetylation is
associated with transcriptional activation and silencing and is
regulated by a balance between the opposing activities of HAT and
HDAC. The inactivation of HAT and activation of HDAC have been
observed in various types of cancers (10). HDAC inhibitors have been reported to
exhibit antitumour effects against various human tumour cells and,
therefore, have become promising candidates for cancer treatment.
Several HDAC inhibitors, such as vorinostat and depsipeptide, have
undergone or are undergoing both phase I and phase II clinical
trials as anticancer agents against haematological malignancies and
solid tumours (12,13). However, there is no reported study
on the implementation of such inhibitors against salivary gland
cancer.
VPA has recently been shown to exhibit activity as
an HDAC inhibitor. VPA has also been reported to prevent the growth
of many human cancers, including neuroblastoma, erythroleukaemia,
acute myelogenous leukemia (11,12),
osteosarcoma (25) and carcinomas
of the breast (8), skin (16), prostate (26), lung (27) and colon (28), suggesting that it can be a useful
agent for the treatment of a wide variety of malignancies. In the
head and neck region, the effect of VPA against squamous cell
carcinomas has been reported (29);
however, its effect against SGC has never been reported. Thus, in
the present study, we investigated the antitumour effects of VPA
against SGCs. It was clearly demonstrated that VPA inhibited the
proliferation of SGC cells in vitro and that VPA treatment
by oral administration effectively inhibited the growth of salivary
gland tumours in xenograft models in vivo. Furthermore, we
observed that VPA induced cell cycle arrest in the G1 phase with
the upregulation of p21 and p27 expression in SGC cells.
Many researchers have investigated the underlying
mechanism of cancer cell growth inhibition induced by VPA acting as
an HDAC inhibitor (7,8,19,25).
Gottlicher et al (30)
demonstrated that VPA relieves HDAC-dependent transcriptional
repression and induces the hyperacetylation of histones in several
types of cancer cells in vitro, as well as reduced tumour
growth and metastasis formation in animal experiments. The authors
indicated that VPA is a powerful HDAC inhibitor and might serve as
an effective drug for cancer therapy. On the other hand, Chen et
al (31) reported that VPA
inhibited the proliferation and invasion of bladder cancer cells by
increasing histone H3 acetylation. In contrast to findings reported
for bladder cancer, VPA had no effect on invasion or migration for
prostate cancer cells. The authors concluded that VPA exerts its
effect in a cancer type-specific manner. In the present study, VPA
inhibited the proliferation of SGC cells in vitro and the
growth of tumours formed by SGC cell inoculation. Although this
antitumour effect of VPA against SGC is not fully understood, VPA
might be able to serve as an effective drug for the treatment of
SGC, acting as an inhibitor of HDAC.
To explore the underlying mechanism of VPA-mediated
cancer cell growth inhibition, we examined the effect of VPA on the
cell cycle phases and distribution of SGC cells treated with
various concentrations of VPA. Our flow cytometric analysis data
indicated that VPA induced the G1 arrest of the cell cycle
progression in SGC cells in a time- and dose-dependent manner.
Numerous reports have indicated that HDAC inhibitors induce cell
cycle arrest, differentiation and apoptosis in many cancer cells by
decreasing the activity of HDAC and leading to the accumulation of
histone hyperacetylation (7,11,14,15).
VPA has also been reported to exert its antitumour effects by
inducing cell cycle arrest, differentiation and apoptosis of cancer
cells. Greenblat et al (32)
demonstrated that VPA activates Notch-1 signalling in human
carcinoid tumour cells in vitro and in vivo, which
results in the inhibition of cell growth via the induction of cell
cycle arrest. VPA is also known to affect several different
signalling pathways, including those of extracellular-regulated
kinase (ERK)-AP-1, protein kinase C (PKC), glycogen synthase
kinase-3 β (GSK-3β) and Wnt. In addition, VPA has recently garnered
attention as a specific inhibitor of HDAC activity (7,19). In
the present study, we did not examine the molecular pathway of VPA
against SGC; however, VPA may act as an effective drug for the
treatment of SGC through these molecular pathways.
Furthermore, to explore the underlying mechanism of
the VPA-mediated G1 arrest of cell cycle progression, we examined
the expression levels of tumour-suppressor genes p21 and p27 in SGC
cells treated with various concentrations of VPA by real-time
RT-PCR. Our data indicated that VPA upregulated the expression of
p21 and p27 in SGC cells in a time-and dose-dependent manner. The
cell cycle progression in mammalian cells is controlled by protein
complexes composed of cyclins and cyclin-dependent kinases (CDKs),
although cell cycle regulation is complex (11,28).
In prostate cancer cells, Sidana et al (34) demonstrated that VPA treatment caused
cell cycle arrest, as determined by the increase in p21 and p27 and
decrease in cyclin D1 expression. p21, a cyclin-dependent kinase
inhibitor, plays an important role in cell cycle progression and
has been shown to be consistently induced by numerous HDAC
inhibitors (11,28). It is well known that p21 is a
transcriptional target of the tumour-suppressor gene p53 and a
downstream effector of p53-dependent cell growth arrest. Induction
of p21 expression may lead to the marked G1 arrest of cancer cells.
In addition, p21 can also bind the proliferation cell nuclear
antigen (PCNA), leading to an inhibition of DNA replication
(33). We previously reported that
a differentiation-inducing drug, vesnarinone, induced histone
hyperacetylation and p21 gene expression, resulting in cell growth
arrest in a human SGC cell line (24). In the present study, VPA induced G1
cell cycle arrest and upregulated p21 expression in SGC cells,
suggesting that VPA may induce histone hyperacetylation. On the
other hand, p27 is a p21-related cyclin-dependent kinase inhibitor
that regulates the progression of cells from the G1 to S phase in
the cell cycle (28).
Downregulation of p27 has been associated with cancer progression
and unfavourable results with respect to several malignancies. It
is well known that reduced expression of p27 is frequently observed
in various cancers, and a lack of p27 is suggested to be due to an
enhancement in its degradation (13). We previously reported that the
upregulation of p27 protein may exert growth-inhibitory effects by
inducing G1 arrest and apoptosis in oral cancer cell lines. p27
also modulates the cell cycle progression and apoptosis of cancer
cells, but its contribution to HDAC inhibitor-induced processes
remains to be fully elucidated.
In the animal experiments performed in the present
study, we administered VPA at a concentration of 0.2 or 0.4% w/v in
drinking water. VPA treatment markedly suppressed the growth of
salivary gland tumours when compared with the growth observed in
the untreated controls, and there was no significant difference
between the 0.2 and 0.4% VPA groups. Shabbeer et al
(8) demonstrated that the
administration of 0.4% w/v VPA in drinking water was effective in
inducing growth arrest, cell death, and senescence in vivo
against prostate cancer. Furthermore, the authors indicated that
the mean plasma level of VPA in mice was 0.4 mM, which is
approximately the level obtained in human patients on VPA. Although
we did not measure the plasma level of VPA in mice, this VPA
concentration (0.4% w/v) poses no significant risk for inducing
toxic effects, including thrombocytopenia and somnolence.
In summary, the present study demonstrated the
possibility of using a new and safe anticancer therapy against SGC,
supporting the use of HDAC as a molecular target for the expression
of tumour-suppressor genes, such as p21 and p27. We provide
evidence that suggests that VPA could contribute to the development
of more effective chemotherapy against SGC. Further studies are
needed to characterise the antitumour activity of VPA in
vivo in other tumour types of SGC for clinical trials.
Abbreviations:
|
VPA
|
valproic acid
|
|
SGC
|
salivary gland cancer
|
|
HDAC
|
histone deacetylase
|
References
|
1
|
Speight PM and Barrett AW: Salivary gland
tumors. Oral Dis. 8:229–240. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bell D and Hanna EY: Salivary gland
cancers: biology and molecular targets for therapy. Curr Oncol Rep.
14:166–174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hunt JL: An update on molecular
diagnostics of squamous and salivary gland tumors of the head and
neck. Arch Pathol Lab Med. 135:602–609. 2011.PubMed/NCBI
|
|
4
|
Laurie SA and Licitra L: Systemic therapy
in the palliative management of advanced salivary gland cancers. J
Clin Oncol. 24:2673–2678. 2006. View Article : Google Scholar
|
|
5
|
Guzzo M, Locati LD, Prott FJ, Gatta G,
McGurk M and Licitra L: Major and minor salivary gland tumors. Crit
Rev Oncol Hematol. 74:134–148. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bensadoun RJ, Blanc-Vincent MP, Chauvel P,
Dassonville O, Gory-Delabaere G and Demard F: Malignant tumours of
the salivary glands. Br J Cancer. 84:42–48. 2001. View Article : Google Scholar
|
|
7
|
Yeow WS, Ziauddin MF, Maxhimer JB, et al:
Potentiation of the anticancer effect of valproic acid, an
antiepileptic agent with histone deacetylase inhibitory activity,
by the kinase inhibitor Staurosporine or its clinically relevant
analogue UCN-01. Br J Cancer. 94:1436–1445. 2006. View Article : Google Scholar
|
|
8
|
Shabbeer S, Kortenhorst MS, Kachhap S,
Galloway N, Rodriguez R and Carducci MA: Multiple molecular
pathways explain the anti-proliferative effect of valproic acid on
prostate cancer cells in vitro and in vivo. Prostate. 67:1099–1110.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosato RR, Almenara JA, Dai Y and Grant S:
Simultaneous activation of the intrinsic and extrinsic pathways by
histone deacetylase (HDAC) inhibitors and tumor necrosis factor
related apoptosis-inducing ligand (TRAIL) synergistically induces
mitochondrial damage and apoptosis in human leukemia cells. Mol
Cancer Ther. 2:1273–1284. 2004.
|
|
10
|
Wolffe AP: Transcription: in tune with the
histones. Cell. 77:13–16. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rosato RR, Almenara JA, Yu C and Grant S:
Evidence of a functional role for p21WAF1/CIP1
down-regulation in synergistic antileukemic interactions between
the histone deacetylase inhibitor sodium butyrate and flavopiridol.
Mol Pharmacol. 65:571–581. 2004.PubMed/NCBI
|
|
12
|
Bug G, Ritter M, Wassmann B, et al:
Clinical trial of valproic acid and all-trans retinoic acid
in patients with poor-risk acute myeloid leukemia. Cancer.
104:2717–2725. 2005.PubMed/NCBI
|
|
13
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schrump DS and Nguyen DM: Targeting the
epigenome for the treatment of thoracic malignancies. Thorac Surg
Clin. 16:367–377. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ruefli AA, Ausserlechner MJ, Bernhard D,
et al: The histone deacetylase inhibitor and chemotherapeutic agent
suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway
characterized by cleavage of Bid and production of reactive oxygen
species. Proc Natl Acad Sci USA. 98:10833–10838. 2001. View Article : Google Scholar
|
|
16
|
Saunders N, Dicker A, Popa C, Jones S and
Dahler A: Histone deacetylase inhibitors as potential anti-skin
cancer agents. Cancer Res. 59:399–404. 1999.PubMed/NCBI
|
|
17
|
De Ruijter AJ, van Gennip AH, Caron HN,
Kemp S and van Kuilenburg AB: Histone deacetylases (HDACs):
characterization of the classical HDAC family. Biochem J.
370:737–749. 2002.PubMed/NCBI
|
|
18
|
Grozinger CM, Hassig CA and Schreiber SL:
Three proteins define a class of human histone deacetylases related
to yeast Hda1p. Proc Natl Acad Sci USA. 96:4868–4873. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blaheta RA and Cinatl J: Anti-tumor
mechanisms of valproate: a novel role for an old drug. Med Res Rev.
22:492–511. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yanagawa T, Hayash Y, Nagamine S, Yoshida
H, Yura Y and Sato M: Generation of ceils with phenotypes of both
intercalated duct-type and myoepithelial cells in human parotid
gland adenocarcinoma clonal cells grown in a thymic nude mice.
Virchows Arch. 51:187–195. 1986. View Article : Google Scholar
|
|
21
|
Shirasuna K, Sato M and Miyazaki T:
Aneoplastic epithelial duct cell line established from an
irradiated human salivary gland. Cancer. 48:745–752. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guan XF, Qiu WL, He RG and Zhou XJ:
Selection of adenoid cystic carcinoma cell clone highly metastatic
to the lung: an experimental study. Int J Oral Maxillofac Surg.
26:116–119. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Uchida D, Begum NM, Aimofti A, Kawamata H,
Yoshida H and Sato M: Frequent downregulation of 14-3-3 σ protein
and hypermethylation of 14-3-3 σ gene in salivary gland
adenoid cystic carcinoma. Br J Cancer. 91:1131–1138. 2004.
|
|
24
|
Uchida D, Onoue T, Begum NM, et al:
Vesnarinone downregulates CXCR4 expression via upregulation of
Kruppel-like factor 2 in oral cancer cells. Mol Cancer. 8:622009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wittenburg LA, Bisson L, Rose BJ, Korch C
and Thamm DH: The histone deacetylase inhibitor varproic acid
sensitizes human and canine osteosarcoma to doxorubicin. Cancer
Chemother Pharmacol. 67:83–92. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang H, Reed CP, Zhang JS, Shridhar V,
Wang L and Smith DI: Carboxypeptidase A3 (CPA3): a novel gene
highly induced by histone deacetylase inhibitors during
differentiation of prostate epithelial cancer cells. Cancer Res.
59:2981–2988. 1999.
|
|
27
|
Sambucetti LC, Fischer DD, Zabludoff S, et
al: Histone deacetylase inhibition selectively alters the activity
and expression of cell cycle proteins leading to specific chromatin
acetylation and antiproliferative effects. J Biol Chem.
274:34940–34947. 1999. View Article : Google Scholar
|
|
28
|
Siavoshian S, Blottiere HM, Cherbut C and
Galmiche JP: Butyrate stimulates cyclin D and p21 and inhibits
cyclin-dependent kinase2 expression in HT-29 colonic epithelial
cells. Biochem Biophys Res Commun. 232:169–172. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Erlich RB, Rickwood D, Coman WB, Saunders
NA and Guminski A: Valproic acid as a therapeutic agent for head
and neck squamous cell carcinomas. Cancer Chemother Pharmacol.
63:381–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gottlicher M, Minucci S, Zhu P, et al:
Valproic acid defines a novel class of HDAC inhibitors inducing
differentiation of transformed cells. EMBO J. 20:6969–6978. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen CL, Sung J, Cohen M, et al: Valproic
acid inhibits invasiveness in bladder cancer but not in prostate
cancer cells. J Pharmacol Exp Ther. 319:533–542. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Greenblatt MS, Bennett WP, Hollstein M and
Harris CC: Mutations in the p53 tumor suppressor gene: clues to
cancer etiology and molecular pathogenesis. Cancer Res.
54:4855–4878. 1994.PubMed/NCBI
|
|
33
|
Vaziri C, Stice L and Faller DV:
Butyrate-induced G1 arrest results from p21-independent disruption
of retinoblastoma protein-mediated signals. Cell Growth Differ.
9:465–474. 1998.PubMed/NCBI
|
|
34
|
Sidana A, Wang M, Shabbeer S, et al:
Mechanism of growth inhibition of prostate cancer xenografts by
valproic acid. J Biomed Biotechnol. 2012:1803632012. View Article : Google Scholar : PubMed/NCBI
|