Introduction
Neuroblastoma, which originates from precursor
neuroblast cells of the sympathetic nervous system, is the most
common solid tumor in children under the age of five. MYCN
oncogene amplification and consequent N-Myc mRNA and protein
over-expression occur in a quarter to a third of tumor tissues and
correlate with poor prognosis in neuroblastoma patients (1,2).
The structurally related Myc oncoproteins, N-Myc and
c-Myc, exert oncogenic effects by modulating gene transcription.
Myc oncoproteins activate gene transcription by direct binding to
Myc-responsive element E-boxes at target gene promoters (3,4), and
repress gene transcription by forming transcriptional repressor
complexes with histone deacetylases (HDACs), including the class I
HDAC1 and HDAC2, the class II HDAC5 as well as the class III SIRT1
and SIRT2, at Sp1-binding sites of target gene promoters (5–9).
Moreover, epigenetic alterations, such as histone H3 lysine 4
(H3K4) tri-methylation at target gene promoter regions, are
considered essential for Myc-mediated transcriptional activation
(10).
JARID1B, also known as KDM5B and PLU1, is a Jumonji
C domain-containing histone demethylase that specifically
demethylates tri- and di-methylated histone H3K4 (11,12).
JARID1B promotes cell differentiation during development, as
JARID1B is essential for neuronal differentiation of embryonic stem
cells (13) and knocking down
JARID1B is associated with reduced levels of
differentiation-associated genes in hematopoietic stem cells
(14). However, JARID1B plays a
dual role in cancer.
In the present, study we identified JARID1B as one
of the genes significantly repressed by Myc, and examined whether
repression of JARID1B contributed to Myc-mediated cancer
phenotypes.
Materials and methods
Cell culture
Neuroblastoma BE(2)-C cells were cultured in
Dulbecco’s modified Eagle’s medium supplemented with 1% L-glutamine
and 10% fetal calf serum. Kelly cells were grown in RPMI-1640
supplemented with 1% L-glutamine and 10% fetal calf serum.
siRNA transfection
Cells were transfected with AllStars negative
control small interfering RNAs (siRNAs), N-Myc siRNAs or JARID1B
siRNAs (Qiagen, Hamburg, Germany) using Lipofectamine®
2000 reagent (Life Technologies, Grand Island, NY, USA) as we
previously described (5–7,9).
Real-time RT-PCR
Following siRNA transfections, RNA was extracted
from cells using PureLink RNA Mini kit (Life Technologies)
according to the manufacturer’s instructions. Synthesis of cDNA
from RNA samples was carried out using M-MLV reverse transcriptase
(Invitrogen, Carlsbad, CA, USA). Real-time RT-PCR was performed
using SYBR-Green PCR Master Mix (Life Technologies) and Applied
Biosystems 7900 as we previously described (5–7,9). The
expression of actin mRNA was employed as loading controls.
Immunoblot analysis
For the analysis of protein expression, cells were
lysed, protein extracted and separated by gel electrophoresis.
After western transfer, membranes were probed with a mouse
anti-N-Myc antibody (1:1,000; Santa Cruz Biotechnology, Santa Cruz,
CA, USA) or a rabbit anti-JARID1B antibody (1:100, Santa Cruz
Biotechnology), followed by a horseradish peroxidase-conjugated
anti-mouse (1:10,000, Santa Cruz Biotechnology) or anti-rabbit
(1:2,000, Santa Cruz Biotechnology) antibody. Protein bands were
visualized with SuperSignal (Pierce, Rockford, IL, USA). The
membranes were lastly re-probed with an anti-actin antibody (Sigma,
St. Louis, MO, USA) as loading controls.
Chromatin immunoprecipitation (ChIP)
assays
ChIP assays were performed with an anti-N-Myc
antibody (Merck Millipore, Billerica, MA, USA) or a control
antibody and PCR with primers targeting negative control region
(−2108 bp to −1946 bp upstream of the transcription start site) or
Sp1-binding site-enriched region (−109 bp to −9 bp upstream of the
transcription start site) of the JARID1B gene. Fold enrichment of
the JARID1B gene core promoter region by the anti-N-Myc antibody
was calculated by dividing the PCR product from the Sp1-binding
site-enriched region by the PCR product from the negative control
region. The sequences of primers targeting the negative control
region were 5′-GCAGCTGAGAATTGGG AAAG-3′ (forward primer) and
5′-TCGGGAGAGCGTTGA CTATT-3′ (reverse primer), and the sequences of
primers targeting the JARID1B core promoter region were 5′-GTGGGG
TGGGACTCTTTTTC-3′ (forward primer) and 5′-CAGCAC CTTGGGCTTTTTC-3′
(reverse primer).
Alamar blue assays
Alamar blue assays were carried out as we previously
described (15). Briefly, cells
were transfected with various siRNAs in 96-well plates. After 72 h,
cells were incubated with Alamar Blue (Invitrogen) for 6 h, and the
plates were read on a microplate reader at 570/595 nm. Results were
calculated according to optical density absorbance units and
expressed as percentage changes in viable cell numbers.
Statistical analysis
The experiments were conducted at least 3 times.
Data are presented as means ± standard error. Differences were
analyzed for significance using ANOVA among groups or unpaired
t-test for two groups. A probability value of 0.05 or less was
considered to indicate a statistically significant difference.
Results
JARID1B gene expression is reactivated
after reduction in Myc or histone deacetylase expression in a range
of cancer cell lines
The interaction between c-Myc protein and JARID1B
protein has been proposed to play an important role in
c-Myc-mediated cell growth (16).
We, therefore, examined the factors which modulated JARID1B gene
expression in published microarray gene expression datasets. As
shown in Table I, knocking down
N-Myc expression with siRNAs significantly upregulated JARID1B mRNA
expression in MYCN oncogene-amplified BE(2)-C neuroblastoma cells
(6,8). Consistently, JARID1B was one of the
genes most markedly upregulated in c-Myc overexpressing KMS11,
MM.1S and OPM1 multiple myeloma cells as well as J-Lat T cells
after treatment with the BET bromodomain inhibitor JQ1, which
exerted anticancer effects by repressing c-Myc gene transcription
(17,18). While histone deacetylases are well
known to form transcriptional repressor complexes with Myc
oncoproteins at Myc target gene promoters, knocking down the
expression of the class I histone deacetylase HDAC2 (6), the class III histone deacetylase SIRT1
(7) or SIRT2 (9) with siRNAs upregulated JARID1B gene
expression in neuroblastoma BE(2)-C cells (Table I). The published microarray gene
expression data suggest that JARID1B is a transcriptional target
gene of Myc oncoproteins and histone deacetylases in a range of
cancer cell lines.
 | Table IJARID1B gene expression is
reactivated after reduction in Myc or histone deacetylase
expression in a range of cancer cell lines. |
Table I
JARID1B gene expression is
reactivated after reduction in Myc or histone deacetylase
expression in a range of cancer cell lines.
| Cell lines | Treatment or siRNA
transfection | Microarray
platform | JARID1B gene
expression | Refs. |
|---|
| BE(2)-C neuroblastoma
cells | N-Myc siRNA | Affymetrix | ↑ | Marshall et al
(6) |
| KMS11, MM.1S, OPM1
multiple myeloma cells | JQ1 treatment | Affymetrix | ↑ | Delmore et al
(17) |
| J-Lat T cells | JQ1 treatment | Affymetrix | ↑ | Banerjee et al
(18) |
| BE()-C neuroblastoma
cells | HDAC2 siRNA | Affymetrix | ↑ | Marshall et al
(6) |
| BE(2)-C neuroblastoma
cells | SIRT2 siRNA | Affymetrix | ↑ | Liu et al
(9) |
| BE(2)-C neuroblastoma
cells | SIRT1 siRNA | Affymetrix | ↑ | Marshall et al
(7) |
N-Myc suppresses JARID1B expression in
neuroblastoma cells
To validate the microarray data that N-Myc
suppressed JARID1B gene expression, we transfected the MYCN
oncogene-amplified BE(2)-C and Kelly human neuroblastoma cells with
control siRNA or two independent siRNAs targeting different regions
of N-Myc mRNA, N-Myc siRNA-1 or N-Myc siRNA-2, followed by
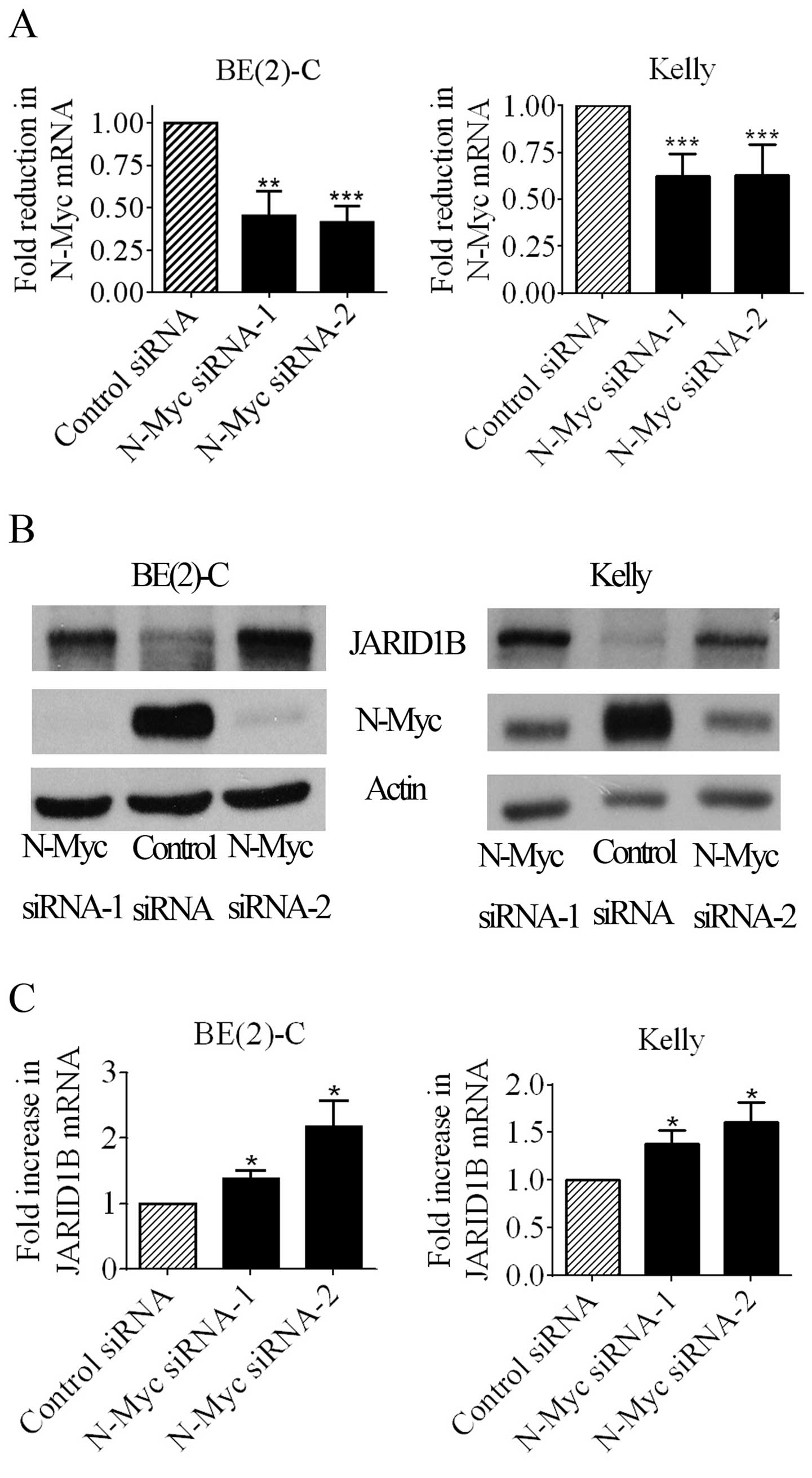
real-time RT-PCR and immunoblot studies of N-Myc and JARID1B. As
shown in Fig. 1A and B, both N-Myc
siRNAs efficiently knocked down N-Myc mRNA and protein expression
in the two neuroblastoma cell lines. Of note, N-Myc siRNA-1 and
N-Myc siRNA-2 increased the expression of JARID1B mRNA (Fig. 1C) and protein (Fig. 1B). The results confirm that N-Myc
represses JARID1B mRNA and protein expression.
N-Myc suppresses JARID1B expression by
direct binding to the JARID1B gene promoter
We previously showed that N-Myc represses gene
transcription by binding to Sp1-binding site-enriched regions of
target gene promoters (5–7,9). To
understand whether N-Myc could directly repress JARID1B gene
transcription, we firstly analyzed transcription factor binding
sites at the JARID1B gene promoter with Gene-Regulation software
(http://www.gene-regulation.com/pub/programs/alibaba2/index.html).
Results showed that Sp1-binding sites were enriched at −179 bp to
−56 bp upstream of the JARID1B gene transcription start site as
well as +4 bp to +873 bp of the JARID1B intron 1 (Fig. 2A). We then examined a c-Myc
chromatin immunoprecipitation-sequencing (ChIP-Seq) dataset, which
was generated by Dr Michael Snyder’s group at Yale University for
the ENCODE/SYDH project (The Encyclopedia of DNA
Elements/Stanford/Yale/USC/Harvard genome project) and deposited at
the University of California Santa Cruz Genome Browser website. As
shown in Fig. 2B, the ChIP-seq data
showed that c-Myc oncoprotein bound to the JARID1B gene promoter
region matching the Sp1-binding site-enriched region in K562
leukemia cells. We next performed ChIP assays with an anti-N-Myc
antibody or a control IgG and PCR with primers targeting −109 bp to
−9 bp upstream of the JARID1B gene transcription start site or a
negative control region. As shown in Fig. 2C, our own ChIP assays showed that
the anti-N-Myc antibody efficiently immunoprecipitated the proximal
JARID1B gene core promoter enriched of Sp1-binding sites in BE(2)-C
neuroblastoma cells. Taken together, the data indicate that N-Myc
reduces JARID1B gene expression by direct binding to the
Sp1-binding site-enriched region of the JARID1B gene core
promoter.
Repression of JARID1B expression reduces
neuroblastoma cell proliferation
To understand whether repression of JARID1B
expression contributed to an N-Myc-regulated cancer phenotype, we
first validated the efficacy of two independent siRNAs targeting
different regions of JARID1B mRNA. RT-PCR and immunoblot analyses
showed that both JARID1B siRNA-1 and JARID1B siRNA-2 knocked down
JARID1B mRNA and protein expression (Fig. 3A and B). We then transfected BE(2)-C
and Kelly cells with control siRNA, N-Myc siRNA-1, N-Myc siRNA-2,
JARID1B siRNA-1 or JARID1B siRNA-2 for 72 h, followed by Alamar
Blue assays. As shown in Fig. 3C,
knocking down N-Myc gene expression with N-Myc siRNA-1 or N-Myc
siRNA-2 reduced the numbers of BE(2)-C and Kelly cells. Similarly,
knocking down JARID1B gene expression with JARID1B siRNA-1 or
JARID1B siRNA-2 also reduced the numbers of BE(2)-C and Kelly cells
(Fig. 3D). It is also noteworthy
that JARID1B siRNAs did not induce neurite outgrowth, an indicator
of neuroblastoma cell differentiation (negative data not shown).
The data suggest that JARID1B induces neuroblastoma cell
proliferation, although N-Myc oncoprotein represses JARID1B
expression.
Discussion
Myc oncoproteins induce tumor initiation and promote
tumor progression by transcriptional repression of tumor suppressor
genes and transcriptional activation of oncogenes, and are
overexpressed in >50% of tumor tissues from the general
population of cancer patients. We previously showed that Myc
oncoproteins repress gene transcription by forming transcriptional
repressor complexes with histone deacetylases at Sp1-binding sites
of target gene promoters (5–8). By
analyzing 35 histone marks after genomic binding by c-Myc, Guccione
et al (10) revealed that
histone H3K4 tri-methylation at Myc-responsive elements of target
gene promoters is a strict prerequisite for Myc-induced
transcriptional activation. However, the mechanism through which
histone H3K4 is tri-methylated during Myc-regulated transcriptional
activation is unknown.
JARID1B specifically demethylates tri- and
di-methylated forms of H3K4. In the present study, we examined
published microarray gene expression data from other groups as well
as our own, and found that JARID1B is one of the genes most
significantly reactivated in a variety of cancer cells after
transfection with siRNAs targeting the histone deacetylase SIRT1,
SIRT2, HDAC2, N-Myc or treatment with the BET bromodomain inhibitor
JQ1, which exerts anticancer effects by repressing N-Myc and c-Myc
gene transcription (17,19,20).
Real-time RT-PCR and immunoblot studies confirmed that N-Myc
reduces JARID1B expression. Bioinformatics analyses show that the
JARID1B gene core promoter is enriched of Sp1-binding sites, and a
publically available ChIP-sequencing dataset shows that c-Myc binds
to the Sp1-binding site-enriched region of the JARID1B gene core
promoter in K562 leukemia cells. Consistently, our own ChIP assays
showed that an anti-N-Myc antibody efficiently immunoprecipitates
the JARID1B gene core promoter enriched in Sp1-binding sites in
neuroblastoma cells. Taken together, the data suggest that N-Myc
represses JARID1B gene transcription by direct binding to the
Sp1-binding site-enriched region of the JARID1B gene promoter, most
likely through recruiting histone deacetylases.
JARID1B has been shown to play a dual role in
cancer. JARID1B expression is significantly downregulated in
advanced and metastatic melanoma tissues, compared with normal
melanocytic nevi (21). Using
JARID1B as a biomarker, a small subpopulation of JARID1B
low-expressing melanoma cells, which give rise to a highly
proliferative progeny, has been characterized. Moreover, knocking
down of JARID1B leads to an initial acceleration of melanoma growth
(22). By contrast, JARID1B is
overexpressed in bladder, lung, breast and prostate tumors at both
mRNA and protein levels (12,23,24),
and knocking down JARID1B expression induces growth inhibition in
cell lines derived from these cancers (12,23,24).
Our results showed that knocking down JARID1B expression reduces
neuroblastoma cell proliferation, further supporting the findings
in bladder, lung, breast and prostate cancer. As N-Myc induces
neuroblastoma cell proliferation and represses JARID1B expression,
our data suggest that N-Myc-mediated transcriptional repression of
JARID1B counterintuitively reduces tumor cell proliferation.
In summary, the present study showed that JARID1B
expression is commonly repressed by Myc oncoproteins and histone
deacetylases in various cancer cell lines, that N-Myc represses
JARID1B expression by direct binding to the Sp1-binding
site-enriched region of the JARID1B gene promoter and that
transcriptional repression of JARID1B counterintuitively reduces
neuroblastoma cell proliferation. While reducing or eliminating
JARID1B protein has been proposed to reduce c-Myc-induced tumors
(16,25), our study suggests that
N-Myc-mediated transcriptional repression of JARID1B
counterintuitively inhibits N-Myc-regulated cell proliferation and
potentially tumorigenesis.
Acknowledgements
This study was supported by the National Health and
Medical Research Council and Cancer Council New South Wales project
grants. T.L. is a recipient of an ARC Future Fellowship. Children’s
Cancer Institute Australia is affiliated with the University of New
South Wales and Sydney Children’s Hospital.
References
|
1
|
Brodeur GM: Neuroblastoma: biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maris JM and Matthay KK: Molecular biology
of neuroblastoma. J Clin Oncol. 17:2264–2279. 1999.PubMed/NCBI
|
|
3
|
Patel JH, Loboda AP, Showe MK, Showe LC
and McMahon SB: Analysis of genomic targets reveals complex
functions of MYC. Nat Rev Cancer. 4:562–568. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eilers M and Eisenman RN: Myc’s broad
reach. Genes Dev. 22:2755–2766. 2008.
|
|
5
|
Liu T, Tee AE, Porro A, et al: Activation
of tissue transglutaminase transcription by histone deacetylase
inhibition as a therapeutic approach for Myc oncogenesis. Proc Natl
Acad Sci USA. 104:18682–18687. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marshall GM, Gherardi S, Xu N, et al:
Transcriptional upregulation of histone deacetylase 2 promotes
Myc-induced oncogenic effects. Oncogene. 29:5957–5968. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marshall GM, Liu PY, Gherardi S, et al:
SIRT1 promotes N-Myc oncogenesis through a positive feedback loop
involving the effects of MKP3 and ERK on N-Myc protein stability.
PLoS Genet. 7:e10021352011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun Y, Liu PY, Scarlett CJ, et al: Histone
deacetylase 5 blocks neuroblastoma cell differentiation by
interacting with N-Myc. Oncogene. Jul 1–2013.(Epub ahead of
print).
|
|
9
|
Liu PY, Xu N, Malyukova A, et al: The
histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death
Differ. 20:503–514. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guccione E, Martinato F, Finocchiaro G, et
al: Myc-binding-site recognition in the human genome is determined
by chromatin context. Nat Cell Biol. 8:764–770. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seward DJ, Cubberley G, Kim S, et al:
Demethylation of trimethylated histone H3 Lys4 in vivo by JARID1
JmjC proteins. Nat Struct Mol Biol. 14:240–242. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiang Y, Zhu Z, Han G, et al: JARID1B is a
histone H3 lysine 4 demethylase up-regulated in prostate cancer.
Proc Natl Acad Sci USA. 104:19226–19231. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schmitz SU, Albert M, Malatesta M, et al:
Jarid1b targets genes regulating development and is involved in
neural differentiation. EMBO J. 30:4586–4600. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cellot S, Hope KJ, Chagraoui J, et al:
RNAi screen identifies Jarid1b as a major regulator of mouse HSC
activity. Blood. 122:1545–1555. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu T, Liu PY, Tee AEL, et al:
Over-expression of clusterin is a resistance factor to the
anti-cancer effect of histone deacetylase inhibitors. Eur J Cancer.
45:1846–1854. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Secombe J, Li L, Carlos L and Eisenman RN:
The Trithorax group protein Lid is a trimethyl histone H3K4
demethylase required for dMyc-induced cell growth. Genes Dev.
21:537–551. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Delmore JE, Issa GC, Lemieux ME, et al:
BET bromodomain inhibition as a therapeutic strategy to target
c-Myc. Cell. 146:904–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Banerjee C, Archin N, Michaels D, et al:
BET bromodomain inhibition as a novel strategy for reactivation of
HIV-1. J Leukoc Biol. 92:1147–1154. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ott CJ, Kopp N, Bird L, et al: BET
bromodomain inhibition targets both c-Myc and IL7R in high-risk
acute lymphoblastic leukemia. Blood. 120:2843–2852. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zuber J, Shi J, Wang E, et al: RNAi screen
identifies Brd4 as a therapeutic target in acute myeloid leukaemia.
Nature. 478:524–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roesch A, Becker B, Meyer S, et al:
Retinoblastoma-binding protein 2-homolog 1: a
retinoblastoma-binding protein downregulated in malignant
melanomas. Mod Pathol. 18:1249–1257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roesch A, Fukunaga-Kalabis M, Schmidt EC,
et al: A temporarily distinct subpopulation of slow-cycling
melanoma cells is required for continuous tumor growth. Cell.
141:583–594. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hayami S, Yoshimatsu M, Veerakumarasivam
A, et al: Overexpression of the JmjC histone demethylase KDM5B in
human carcinogenesis: involvement in the proliferation of cancer
cells through the E2F/RB pathway. Mol Cancer. 9:592010. View Article : Google Scholar
|
|
24
|
Barrett A, Madsen B, Copier J, et al:
PLU-1 nuclear protein, which is upregulated in breast cancer, shows
restricted expression in normal human adult tissues: a new
cancer/testis antigen? Int J Cancer. 101:581–588. 2002. View Article : Google Scholar
|
|
25
|
Secombe J and Eisenman RN: The function
and regulation of the JARID1 family of histone H3 lysine 4
demethylases: the Myc connection. Cell Cycle. 6:1324–1328. 2007.
View Article : Google Scholar : PubMed/NCBI
|