Introduction
Pancreatic cancer, which is one of the most
aggressive and lethal cancers worldwide, is highly resistant to
chemotherapy (1). Even systemic
therapy with gemcitabine, a current first-line treatment for
advanced pancreatic cancer, offers only modest benefit due to
pre-existing or acquired chemoresistance (2,3).
Furthermore, clinical studies indicate that only 12% of patients
with advanced pancreatic cancer exhibit a response to gemcitabine
(4). The poor response rate
suggests that pancreatic cancer either rapidly develops or has
intrinsic gemcitabine chemoresistance. The mechanisms by which
chemoresistance arises in pancreatic cancer are unknown; thus a
better understanding of how resistance arises and what molecular
alterations cause or correlate with resistance is likely to lead to
novel therapeutic strategies for pancreatic cancer.
Hypoxia is an environmental stimulus that plays a
key role in the development and progression of cancer. Tumoral
hypoxia or expression of hypoxia-inducible factor-1 (HIF-1) has
been linked to an aggressive phenotype which correlates with a poor
response to chemotherapy and a worse overall survival of cancer
patients (5,6). HIF-1 is a heterodimeric protein
consisting of a constitutively expressed subunit, HIF-1β, and an
oxygen-sensitive inducible subunit, HIF-1α. Under normoxia, the
HIF-1α protein is hydroxylated by a family of oxygen-dependent
prolyl hydroxylases (PHD1–3); this targets it for
polyubiquitination by a protein complex containing von
Hippel-Lindau protein (pVHL) and then degradation (7). Under hypoxic conditions, prolyl
hydroxylases are inactivated and HIF-1α degradation is blocked;
this allows HIF-1α to accumulate and associate with HIF-1β to form
a functional transcription complex that triggers the transcription
of a host of hypoxia-inducible genes (7).
Epithelial-to-mesenchymal transition (EMT) is the
process by which adherent epithelial cells convert to motile
mesenchymal cells and is essential in embryonic development. EMT is
now known to also occur in a variety of diseases including the
progression of cancer (8). In a
previous study, evidence was provided suggesting that moderate
hypoxic conditions can trigger, acting as an independent factor, an
EMT program leading different human cancer cells to significantly
increased invasiveness (9).
Detailed examinations of the multiple facets of the EMT program
have revealed its involvement in more than just invasion and
metastasis; previous studies show that the phenotype of EMT is
associated with chemoresistance in a diverse array of solid tumors
(10–17).
Nuclear factor-κB (NF-κB) is a ubiquitous
transcription factor regulated by numerous stimuli including
hypoxia, cytokines and chemotherapeutic drugs, and has recently
emerged as a target for cancer. NF-κB is constitutively activated
in most (>70%) human pancreatic cancer cells and in primary
tumor specimens, but not in normal pancreatic tissues or
non-tumorigenic cell lines (18–20).
Previous studies have shown that hypoxia activates NF-κB (21,22),
and induces the resistance of pancreatic cancer cells to
gemcitabine (23), whereas NF-κB
can also regulate HIF-1 (24,25).
Some studies have also reported that activation of NF-κB is closely
involved in the progression of EMT (26–29).
Recently, we reported that dihydroartemisinin or small interfering
RNA (siRNA) inactivates NF-κB and potentiates the antitumor effect
of gemcitabine on pancreatic cancer both in vitro and in
vivo (30,31). Compared with HIF-1α, the regulation
of NF-κB and EMT during hypoxia has been less studied, and although
the phenomenon was previously observed, the molecular mechanisms
involved remain unclear. Thus, we sought to determine the role of
the HIF-1α and NF-κB loop in the hypoxic microenvironment in
pancreatic cancer. Therefore, the present study was designed to
investigate whether downregulation of the p65 subunit of NF-κB or
HIF-1α by siRNA may reverse the EMT phenotype and inhibit the
proliferation and induce the apoptosis of pancreatic cancer cells
(PANC-1, BxPC3) under hypoxic conditions and enhance the efficacy
of gemcitabine to treat pancreatic cancer.
Materials and methods
Cell lines and reagents
The human pancreatic cancer cell lines BxPC-3 and
PANC-1 were obtained from the American Type Culture Collection
(Rockville, MD, USA). Both cell lines were routinely cultured at
37°C in RPMI-1640 medium supplemented with 10% fetal calf serum,
penicillin (100 U/ml) and streptomycin (100 μg/ml) in an incubator
with 95% air and 5% CO2. Hypoxic control cells were
incubated under the same conditions but in a hypoxic incubator
(Precision Scientific, Winchester, VA, USA) with 1% O2,
5% CO2 and 94% N2. The antibodies against
NF-κB(p65), SNAI1(H-130), TWIST(H-81) and β-actin were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The
HIF-1α Ab was purchased from Abcam Inc. (Cambridge, MA, USA). The
Vimentin, E-cadherin and N-cadherin Abs were purchased from
ZSGB-BIO Inc. (Beijing, China). Annexin V-FITC and propidium iodide
(PI) were purchased from Sigma Inc. (Beijing, China). Gemcitabine
was purchased from Lily France (Fegersheim, France).
Preparation of siRNAs and
transfection
To knock down the HIF-1α and NF-κB p65 subunit,
synthesized siRNA duplexes were obtained from Invitrogen. A
double-stranded siRNA (p65 siRNA) (sense, 5′-GCCCUA UCCCUU UACGUC
A-3′ and antisense 5′-UGACGU AAAGGG AUAGGG C-3′) with two
introduced thymidine residues at the 3′ end, which encode amino
acid residues 347 and 353 of the NF-κB p65 subunit was designed to
target the NF-κB p65 subunit as described previously (31). Based on a previous report (32), the sequences of two oligonucleotides
targeting HIF-1α were as follows; sense, 5′-GGGUAA AGAACA AAACAC
A-3′ and antisense 5′-UGUGUU UUGUUC UUUACC C-3′. BxPC-3 and PANC-1
cells were grown to 50% confluency in 6- or 96-well plates, and
transfected with the siRNAs in serum-free medium without antibiotic
supplements using Lipofectamine™ 2000 (Invitrogen). Cells were
allowed to stabilize for 48 h before being used in the experiments.
Silencing of protein expression was confirmed by western blot
analysis.
Assessment of cellular morphological
changes and determination of cell viability
Cellular morphology was evaluated using
phase-contrast microscopy, and images were captured with a
computer-imaging system (Olympus Q-Color 3RTV camera). Cell
viability was determined by the Cell Counting Kit-8 assay (Dojin
Laboratory, Kumamoto, Japan) and crystal violet assay.
Cell Counting Kit-8 assay
BxPC-3 (3×103/well) and PANC-1
(4×103/well) cells were split into a 96-well plate and
incubated overnight in a humidified CO2 incubator (95%
air and 5% CO2) to allow the cells to adhere and assume
a healthy condition, and were then exposed to gradient doses of
gemcitabine (0–200 μmol/l) for 48 h under hypoxic and normoxic
conditions. The cells were then incubated with WST-8 solution at
37°C for 1 h, and the absorbance at 450 nm was measured on a
microplate reader (MPR-A4i; Tosoh Corporation, Tokyo, Japan). The
cells cultured in RPMI-1640 medium served as the control. The cell
viability index was calculated according to the formula:
Experimental OD value/control OD value × 100%. All experiments were
carried out in triplicate and repeated thrice.
Crystal violet assay
The cells (5×104/well) were seeded into
6-well plates and cultured overnight. Then the medium was replaced
with complete culture medium containing gemcitabine (10 μmol/l for
PANC-1 and 500 nmol/l for BxPC-3 cells) for an additional 48 h
under hypoxic and normoxic conditions. Cells were then washed twice
with pre-warmed PBS, and then the cells remaining were stained for
2 h with a crystal violet solution (0.5% crystal violet, 20%
methanol). After removal of the crystal violet solution, the plates
were washed three times by immersion in a beaker filled with tap
water. Plates were left to dry at 37°C, and then the images were
captured by camera.
Detection of cell apoptosis
After the designated treatment, BxPC-3 and PANC-1
cells were washed, harvested and counted. Cells (1×106)
were resuspended in 100 μl binding buffer, before 5 μl of Annexin V
and 5 μl of PI were added, and incubated for 15 min at room
temperature in the dark, according to the manufacturer’s
instructions (Biosea, China). The apoptosis rate (%) was measured
in a cytometer (Epics Altra II; Beckman Coulter, USA). Cells were
viewed under a laser scanning confocal microscope (LSM-510; Carl
Zeiss Jena GmbH, Jena, Germany) to visualize those that had
undergone apoptosis. The experiment was repeated thrice.
Electrophoretic mobility shift assay
(EMSA)
EMSA was performed by incubating 10 μg of nuclear
protein, prepared according to a method described previously
(30), with Gel Shift Binding
buffer and 1 μg of poly(deoxyinosinic-deoxycytidylic acid) at 4°C
for 30 min, and then mixed with biotin-labeled oligonucleotide
bio-NF-κB probe (5′-AGTTGA GGGGAC TTTCCC AGGC-3′) at room
temperature for 20 min, according to the manufacturer’s
instructions (Viagene, Beijing, China). The resulting DNA-protein
complex was separated from the free oligonucleotide on a 4%
polyacrylamide gel containing 0.25× TBE (Tris/borate/EDTA) buffer.
The dried gels were visualized with a Cool Imager imaging system
(IMGR002), and radioactive bands were quantified using Scion Image
software.
Western blotting
The methodology was described previously (30). Briefly, 5×105 cells were
sonicated in RIPA buffer and homogenized. Debris was removed by
centrifugation at 12,000 × g for 10 min at 4°C. The samples
containing 50 μg protein were electrophoresed on polyacrylamide SDS
gels, and transferred to polyvinylidene difluoride (PVDF)
membranes. The membranes were blocked with 3% BSA, incubated with
primary antibodies, and subsequently with alkaline
phosphatase-conjugated secondary antibody. They were developed with
5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium
(Tiangen Biotech Co. Ltd., Beijing, China). Blots were also stained
with anti-β-actin Ab as the internal control for the amounts of
target proteins.
Statistical analysis
The results are expressed as mean values ± standard
deviation, and a Student’s t-test was used to evaluate statistical
significance. A value of <0.05 (P<0.05) was used to indicate
statistical significance.
Results
Morphological and cell biological changes
characteristic of EMT under hypoxic conditions
In preliminary experiments, 65–70% subconfluent
pancreatic cancer cells (PANC-1, BxPC3) were exposed to hypoxic
conditions (1% O2, 5% CO2 and 94%
N2) up to 48 h. As reported previously (9), PANC-1 cells exposed to hypoxia up to
48 h underwent typical morphological and cellular changes of EMT;
PANC-1 cells started to loose cell contacts, scattered from cell
clusters and acquired a spindle-shaped and fibroblast-like
phenotype. In the BxPC3 cell lines, however, morphological and
cellular changes did not significantly differ (Fig. 1A).
 | Figure 1Hypoxia induces an EMT-like
fibroblastoid phenotype in pancreatic cancer cells. (A) Phase
contrast analysis (original magnification, ×100) of morphological
changes detected in human pancreatic cancer cell lines under
normoxic (N, 21% O2) or hypoxic (H, 1% O2)
conditions. Representative images were captured after a 48-h
incubation. (B) Western blot analysis of the expression of
E-cadherin, Vimentin, N-cadherin, Snail and Twist from respective
cell homogenate, with β-actin used as a protein loading control.
Bottom panels: relative intensity of data presented in the top
panels, compared with the protein loading control.
*P<0.05, significant difference in the relative
intensity when compared to a normoxic condition (N, 21%
O2). EMT, epithelial-to-mesenchymal transition. |
To further confirm whether pancreatic cancer cells
underwent EMT when exposed to hypoxia, we also determined the
expression of markers of epithelial and mesenchymal phenotypes by
western blotting. Induction of EMT was previously demonstrated in
both cell lines after hypoxia, as shown by a shift in expression of
epithelial markers (E-cadherin) to mesenchymal markers (Vimentin
and N-cadherin) and various transcription factors of EMT, such as
Snail and Twist, were overexpressed (33,34).
We found that pancreatic cancer cells (PANC-1, BxPC3) after
exposure to hypoxia for 48 h had reduced expression of E-cadherin
and increased expression of Vimentin, N-cadherin, Snail and Twist
when compared with cells under a normoxic condition (Fig. 1B).
Hypoxia induces HIF-1α and NF-κB
hyperexpression and decreases the susceptibility to gemcitabine of
pancreatic cancer cells
Studies have previously shown that hypoxia results
in activation of NF-κB (21,22)
and EMT. Thus, we sought to determine whether the EMT observed in
pancreatic cancer cells (PANC-1, BxPC3) was attributable to
heightened NF-κB activity. First, to verify the expression of NF-κB
and HIF-1α during hypoxia, pancreatic cancer cells were incubated
under hypoxic conditions for different periods of time (24, 48 and
72 h). After each indicated incubation period, the cells were
collected, and total and nuclear proteins were extracted. HIF-1α
and NF-κB were analyzed by western blot analysis (Fig. 2A). The NF-κB DNA binding activity
was also determined by EMSA (Fig.
2B). Expression levels of HIF-1α and NF-κB proteins were
increased 24 h after the initiation of hypoxic treatment and
reached a maximum by 72 h. NF-κB DNA binding activity was also
increased 24 h after initiation of hypoxia and reached maximum
activity by 72 h. These data demonstrated that the expression
levels of HIF-1α and NF-κB were increased under hypoxia when
compared to these levels under normoxic conditions in a
time-dependent manner. Moreover, some studies have previously shown
that EMT is a key step of drug resistance in a number of different
tumors (10–17). Thus, we sought to examine the
effects of EMT induced by hypoxia on the response to gemcitabine by
pancreatic cancer cells. Pancreatic cancer cells, PANC-1 and
BxPC-3, were divided into two groups (N/normoxia and H/hypoxia) and
exposed to gradient doses of gemcitabine (0–200 μmol/l) for 48 h.
Cell Counting Kit-8 assay was used to assess the cell viability
rate. Data are shown in Fig. 2C.
When treated with concentrations of gemcitabine >100 nmol/l,
hypoxia cells showed a significantly higher cell viability rate
when compared with the normoxia cells (P<0.05) (Fig. 2C). These results were further
confirmed by crystal violet assay (Fig.
2D). When treated with gemcitabine (10 μmol/l for PANC-1 and
500 nmol/l for BxPC-3) for 48 h, cells incubated under hypoxic
conditions showed a significantly higher cell viability rate than
that under a normoxic condition. Together, these findings validate
the involvement of hypoxia in the activation of NF-κB and HIF-1α
stabilization and chemoresistance to gemcitabine in pancreatic
cancer cells.
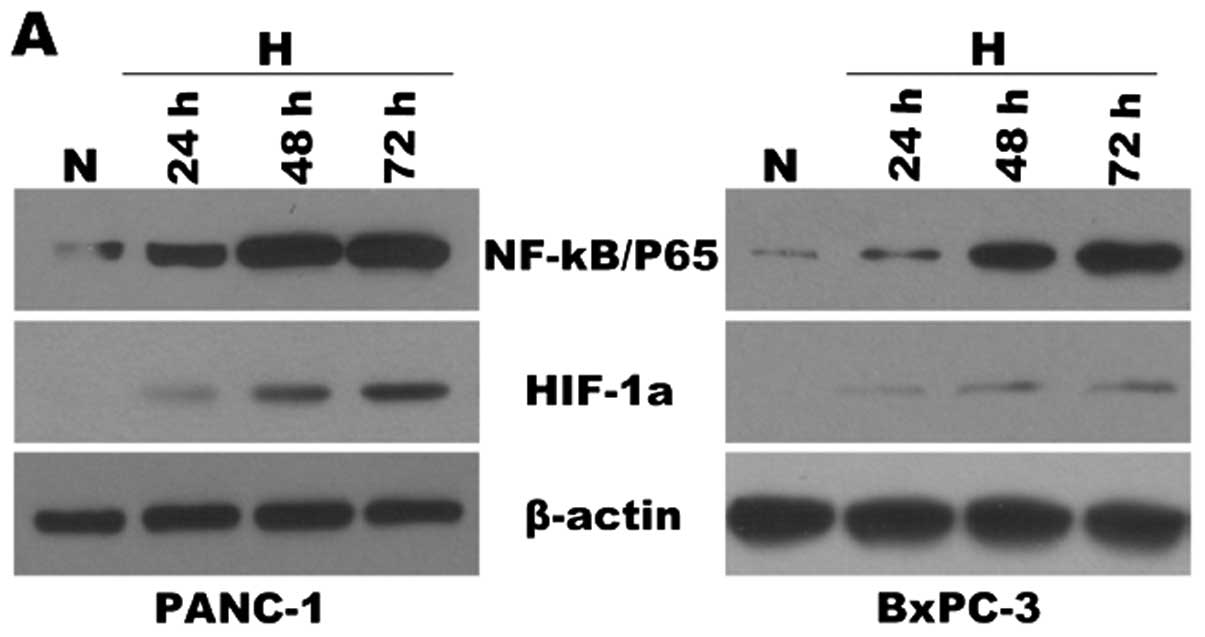 | Figure 2Hypoxia induces HIF-1α and NF-κB
overrexpression and decreases the susceptibility to gemcitabine of
pancreatic cancer cells. PANC-1 and BxPC-3 cells were cultured
under normoxic (N, 21% O2) or hypoxic conditions (H, 1%
O2) for various time periods (24, 48 and 72 h). After
each indicated incubation period, the cells were collected. Nuclear
extracts and total protein extracts were prepared. (A) HIF-1α and
NF-κB(p65) were analyzed by western blot analysis from respective
cell homogenates, with β-actin used as the protein loading control.
(B) Electrophoretic mobility shift assay (EMSA) for NF-κB DNA
binding activity of the respective nuclear extracts. Competitive
assay confirmed the specificity of NF-κB binding to the DNA. Lane
1, control; lane 2, excess unlabeled (‘cold’) NF-κB
oligonucleotide. Bottom panel: densitometric quantification of the
data presented in the top panel. *P<0.05 and
†P<0.01, compared with normoxia. (C) PANC-1 and
BxPC-3 cells were divided into two groups (normoxia/N and
hypoxia/H) and then exposed to gradient doses of gemcitabine (0–200
μmol/l) for 48 h. Cell Counting Kit-8 assay was used to assess the
cell viability rate. *P<0.05, significantly higher
cell viability rate when compared to normoxia (N, 21%
O2). (D) The proliferation of cells was also measured by
crystal violet assay. Images were selected as representative scenes
from three independent experiments. HIF-1α, hypoxia-inducible
factor-1α; NF-κB, nuclear factor-κB. |
Effects of HIF-1α and NF-κB on EMT under
hypoxic conditions
To test the effects of HIF-1α and NF-κB on the
expression of specific EMT markers under hypoxic conditions, we
knocked down NF-κB using siRNA in pancreatic cancer cells (PANC-1,
BxPC3). As shown in Fig. 3A, the
cells manifested reduced E-cadherin expression and increased
Vimentin, N-cadherin, Snail and Twist expression under hypoxic
conditions when compared with cells under normoxic conditions.
Similar results were obtained when we silenced HIF-1α using siRNA.
It has been shown that downregulation of NF-κB or HIF-1α is
associated with EMT. Furthermore, cells that have undergone EMT
tend to exhibit greater chemoresistance. The next question that
remained to be addressed was whether HIF-1α and NF-κB contributed
to the formation of the resistance. We examined these properties in
pancreatic cancer cells (PANC-1, BxPC3) with flow cytometry to
confirm the role of HIF-1α and NF-κB in cells that underwent EMT
resisting the apoptosis induced by gemcitabine. To test this,
BxPC-3 and PANC-1 cells were transfected with HIF-1α siRNA and
NF-κB(p65) siRNA respectively, and the transfectants were treated
with gemcitabine for 48 h under normoxic and hypoxic conditions.
NF-κB(p65) siRNA or HIF-1α siRNA both abrogated the increase in
NF-κB DNA binding activity induced by hypoxia when compared to the
activity under normoxia and reversed the resistance of pancreatic
cancer cells to gemcitabine. Furthermore, we also performed western
blotting and EMSA (Fig. 3B) to
determine the correlation between the expression of HIF-1α and
NF-κB under hypoxic conditions. As shown in Fig. 3A, NF-κB(p65) siRNA had an effect on
the expression of HIF-1α when compared to the control under hypoxic
conditions, and vice versa. The data indicated that HIF-1α and
NF-κB may affect each other and both may reverse EMT and the
resistance of pancreatic cancer cells to gemcitabine induced by
hypoxia.
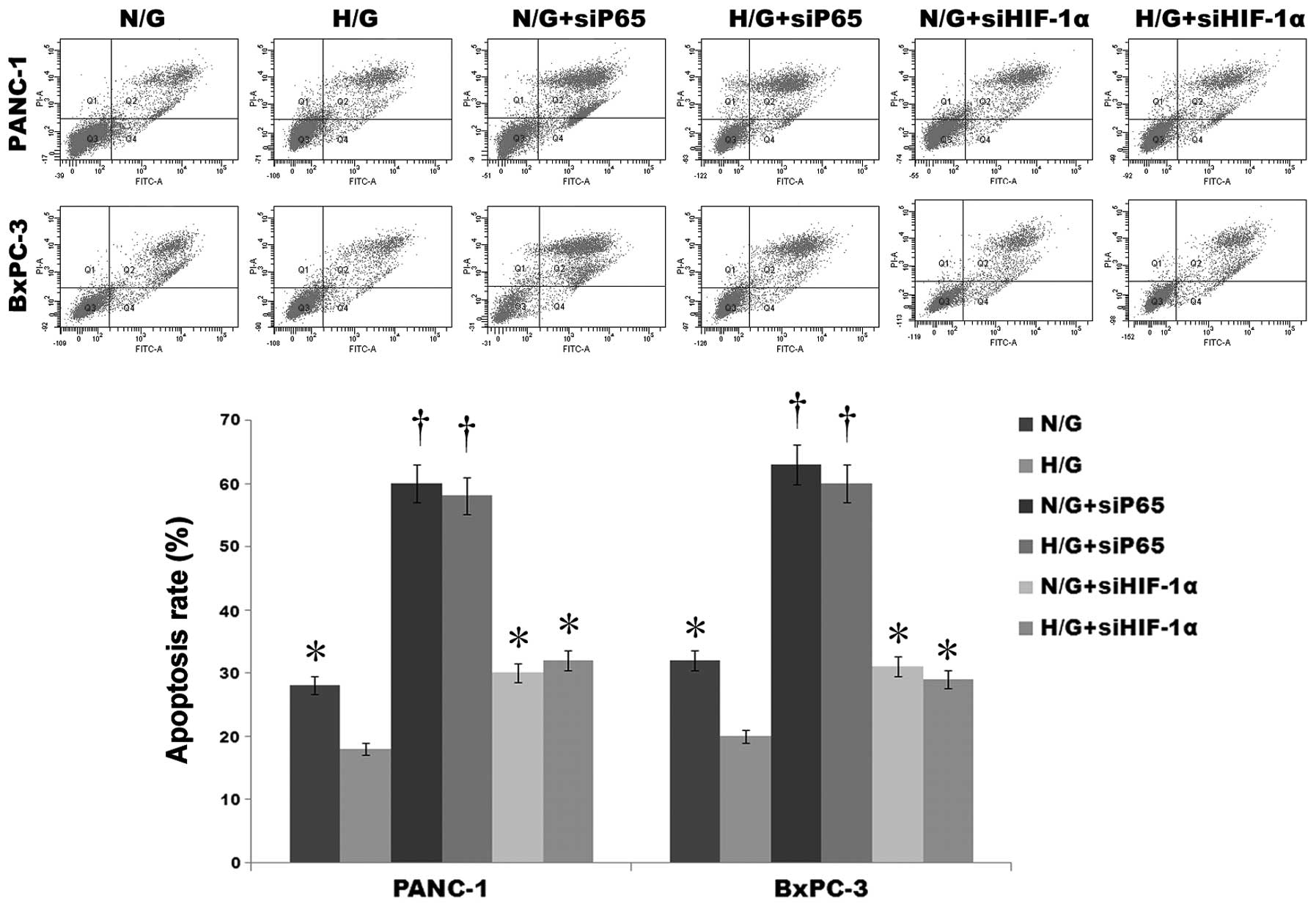
As designated, PANC-1 and BxPC-3 cells were treated
with gemcitabine (G/10 μmol/l and 500 nmol/l, respectively), or a
combination with HIF-1α siRNA (G+si HIF-1α) or NF-κB(p65) siRNA
(G+siP65) for 48 h under hypoxic (H) and normoxic (N) conditions,
before being stained with Annexin V/PI for flow cytometric
determination of the apoptotic rate.
As shown in Fig. 4,
the apoptotic rate was increased significantly when cells were
treated with gemcitabine under normoxic conditions (N/G) when
compared with the rate in cells treated with gemcitabine under
hypoxic conditions (H/G) (both P<0.05). This result suggests
that the apoptosis induced by gemcitabine can be resisted by
hypoxia.
Furthermore, our data showed that the apoptosis rate
was increased significantly when cells were treated with a
combination of gemcitabine and HIF-1α siRNA under hypoxic
conditions (H/G+siHIF-1α) or NF-κB(p65) siRNA under hypoxic
conditions (H/G+siP65) compared with treatment with gemcitabine
alone under hypoxic conditions (H/G) for 48 h (P<0.05,
P<0.01, respectively). Meanwhile, our data also showed that the
apoptosis rate was not significantly altered when the cells were
treated with the combination of gemcitabine and HIF-1α siRNA
(H/G+siHIF-1α) or NF-κB(p65) siRNA (H/G+siP65) under hypoxic
conditions when compared with the apoptosis rate under normoxic
conditions (N/G+siHIF-1α, N/G+siP65) for 48 h. These findings
validate that the involvement of hypoxia in the resistance to
apoptosis induced by gemcitabine was mainly attributable to HIF-1α
and NF-κB hyperactivity.
Together, our results suggest that hypoxia-induced
EMT and resistance to gemcitabine were mainly attributable to
HIF-1α and NF-κB hyperactivity.
Discussion
A hypoxic microenvironment is commonly found in the
central region of solid tumors. Since hypoxia in tumors is
associated with poor prognosis, resistance to chemotherapy and
radiation therapy, and increased metastatic potential, targeting
hypoxia response pathways is of potential therapeutic value
(5). In recent years, it has become
increasingly clear that EMT plays important roles in the
progression of cancer and is also responsible for the resistant
phenotype of cancer cells to conventional chemotherapeutics
(10–17). Induction of EMT has been previously
demonstrated following hypoxia, as shown by a shift in expression
of epithelial markers (E-cadherin) to mesenchymal markers (Vimentin
and N-cadherin) (33–36). A number of factors including the
zinc finger Snail and several basic helix-loop-helix factors such
as Twist that transcriptionally repress E-cadherin have emerged as
potent EMT drivers during normal development and cancer (37,38).
Overexpression of Twist was found to result in an increase in
N-cadherin, which leads to a further decrease in E-cadherin
expression (33,39). In the present study, we found that
when pancreatic cancer cells were exposed to hypoxic conditions for
up to 48 h, PANC-1 cells underwent typical EMT morphological and
cellular changes. These cells lost cell contact, scattered from
cell clusters and acquired a spindle-shaped and fibroblast-like
phenotype. In the BxPC3 cell line, however, morphological and
cellular changes did not differ significantly. Thus, BxPC3 cells
were more sensitive to chemotherapeutics than PANC-1 cells. The
loss of cellular polarity and homotypic adhesion are major
components of EMT. One study reported that drug-resistant cells
displayed a ‘more mesenchymal’ phenotype (12). To further confirm whether pancreatic
cancer cells undergo EMT when exposed to hypoxia, we determined the
expression of markers of epithelial and mesenchymal phenotypes by
western blotting. We found that pancreatic cancer cells (PANC-1,
BxPC-3) after exposure to hypoxia for 48 h had reduced expression
of E-cadherin and increased expression of Vimentin, N-cadherin,
Snail and Twist when compared with the levels in cells under a
normoxic condition. Furthermore, the pancreatic cancer cells that
underwent EMT exhibited a resistant phenotype to conventional
chemotherapeutics. Our study also confirmed that hypoxia was
responsible for chemoresistance to gemcitabine in pancreatic cancer
cells that underwent EMT. In the present study, the process of EMT
in pancreatic cancer cells under a hypoxic condition was
characterized by N-cadherin, Vimentin, Snail and Twist
overexpression and E-cadherin suppression, striking morphological
changes to the mesenchymal phenotype and a drug resistance to
gemcitabin.
Although different factors are involved in the
induction of EMT, studies have previously indicated that HIF-1α
(33) and NF-κB (26–29)
have a critical role in EMT. It was previously reported that HIF-1α
may regulate NF-κB expression (21,22,40,41).
Other studies reported that NF-κB transcriptionally induces HIF-1α
expression (24,25). Therefore, the correlation between
NF-κB and HIF-1 is not fully defined. In our experiments, EMSA
performed with the NF-κB probe in pancreatic cancer cells
demonstrated that the DNA binding activity of NF-κB was increased
as well as the expression of NF-κB(p65) and HIF-1α protein in
hypoxic cells in a time-dependent manner. Our data also
demonstrated that the overexpression of NF-κB(p65) protein and
NF-κB DNA binding activity were inhibited by HIF-1α siRNA, and vice
versa. Expression of HIF-1α protein was also inhibited by
NF-κB(p65) siRNA. It is well known that EMT characteristics must be
reversed to overcome drug resistance, which may lead to the
sensitization of drug-resistant cancer cells to conventional
chemotherapeutic agents. The results presented in the present study
showed that HIF-1α siRNA and NF-κB(p65) siRNA, respectively, may
reverse EMT and increase the sensitivity of pancreatic cancer cells
to gemcitabine. The complete results of the present study indicated
that the EMT program attributable to hypoxia was driven by a
biochemical loop, NF-κB and HIF-1, and these two principal
transcription factors can regulate each other in this process.
In conclusion, these exciting results should provide
incentives for further investigation and optimization in
establishing the mechanistic role of the HIF-1α and NF-κB loop in
the reversal of EMT characteristics and drug resistance and their
utility in the clinical setting for the treatment of pancreatic
cancer for which there is no effective and curative therapy.
Acknowledgements
This work was supported in part by grants from the
Natural Science Foundation of Heilongjiang Province, China (H201373
to Z.-X.C.), Jiamusi University Youth Foundation (q2013-024 to
Z.-X.C.). China Postdoctoral Science Foundation, China (2012M520769
to D.-W.W.) and Hei Long Jiang Postdoctoral Foundation, China
(LBH-Z12201 to D.-W.W.). The funders had no role in study design,
data collection and analysis, decision to publish, or preparation
of the manuscript.
References
|
1
|
Beger HG, Rau B, Gansauge F, Poch B and
Link KH: Treatment of pancreatic cancer: challenge of the facts.
World J Surg. 27:1075–1084. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kullmann F, Hollerbach S, Dollinger MM,
Harder J, Fuchs M, Messmann H, Trojan J, Gäbele E, Hinke A,
Hollerbach C and Endlicher E: Cetuximab plus
gemcitabine/oxaliplatin (GEMOXCET) in first-line metastatic
pancreatic cancer: a multicentre phase II study. Br J Cancer.
100:1032–1036. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Herrmann R, Bodoky G, Ruhstaller T,
Glimelius B, Bajetta E, Schüller J, Saletti P, Bauer J, Figer A,
Pestalozzi B, Köhne CH, Mingrone W, Stemmer SM, Tàmas K, Kornek GV,
Koeberle D, Cina S, Bernhard J, Dietrich D and Scheithauer W; Swiss
Group for Clinical Cancer Research; Central European Cooperative
Oncology Group. Gemcitabine plus capecitabine compared with
gemcitabine alone in advanced pancreatic cancer: a randomized,
multicenter, phase III trial of the Swiss Group for Clinical Cancer
Research and the Central European Cooperative Oncology Group. J
Clin Oncol. 25:2212–2217. 2007. View Article : Google Scholar
|
|
4
|
Bergman AM, Pinedo HM and Peters GJ:
Determinants of resistance to 2′,2′-difluorodeoxycytidine
(gemcitabine). Drug Resist Updat. 5:19–33. 2002.
|
|
5
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maxwell PH: The HIF pathway in cancer.
Semin Cell Dev Biol. 16:523–530. 2005. View Article : Google Scholar
|
|
7
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar
|
|
8
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cannito S, Novo E, Compagnone A, Valfrè di
Bonzo L, Busletta C, Zamara E, Paternostro C, Povero D, Bandino A,
Bozzo F, Cravanzola C, Bravoco V, Colombatto S and Parola M: Redox
mechanisms switch on hypoxia-dependent epithelial-mesenchymal
transition in cancer cells. Carcinogenesis. 29:2267–2278. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shah AN, Summy JM, Zhang J, Park SI,
Parikh NU and Gallick GE: Development and characterization of
gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol.
14:3629–3637. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A,
Azmi AS, Ali S, Abbruzzese JL, Gallick GE and Sarkar FH:
Acquisition of epithelial-mesenchymal transition phenotype of
gemcitabine-resistant pancreatic cancer cells is linked with
activation of the Notch signaling pathway. Cancer Res.
69:2400–2407. 2009. View Article : Google Scholar
|
|
12
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ and Choi W: Epithelial to mesenchymal transition contributes to
drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hiscox S, Jiang WG, Obermeier K, Taylor K,
Morgan L, Burmi R, Barrow D and Nicholson RI: Tamoxifen resistance
in MCF7 cells promotes EMT-like behaviour and involves modulation
of β-catenin phosphorylation. Int J Cancer. 118:290–301.
2006.PubMed/NCBI
|
|
14
|
Kajiyama H, Shibata K, Terauchi M,
Yamashita M, Ino K, Nawa A and Kikkawa F: Chemoresistance to
paclitaxel induces epithelial-mesenchymal transition and enhances
metastatic potential for epithelial ovarian carcinoma cells. Int J
Oncol. 31:277–283. 2007.
|
|
15
|
Yang AD, Fan F, Camp ER, van Buren G, Liu
W, Somcio R, Gray MJ, Cheng H, Hoff PM and Ellis LM: Chronic
oxaliplatin resistance induces epithelial-to-mesenchymal transition
in colorectal cancer cell lines. Clin Cancer Res. 12:4147–4153.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fuchs BC, Fujii T, Dorfman JD, Goodwin JM,
Zhu AX, Lanuti M and Tanabe KK: Epithelial-to-mesenchymal
transition and integrin-linked kinase mediate sensitivity to
epidermal growth factor receptor inhibition in human hepatoma
cells. Cancer Res. 68:2391–2399. 2008. View Article : Google Scholar
|
|
17
|
Yauch RL, Januario T, Eberhard DA, Cavet
G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S,
Modrusan Z, Lin CY, O’Neill V and Amler LC: Epithelial versus
mesenchymal phenotype determines in vitro sensitivity and predicts
clinical activity of erlotinib in lung cancer patients. Clin Cancer
Res. 11:8686–8698. 2005. View Article : Google Scholar
|
|
18
|
Wang W, Abbruzzese JL, Evans DB, Larry L,
Cleary KR and Chiao PJ: The nuclear factor-κB RelA transcription
factor is constitutively activated in human pancreatic
adenocarcinoma cells. Clin Cancer Res. 5:119–127. 1999.
|
|
19
|
Liptay S, Weber CK, Ludwig L, Wagner M,
Adler G and Schmid RM: Mitogenic and antiapoptotic role of
constitutive NF-κB/relactivity in pancreatic cancer. Int J Cancer.
105:735–746. 2003.PubMed/NCBI
|
|
20
|
Chandler NM, Canete JJ and Callery MP:
Increased expression of NF-κB subunits in human pancreatic cancer
cells. J Surg Res. 118:9–14. 2004.
|
|
21
|
Chandel NS, Trzyna WC, McClintock DS and
Schumacker PT: Role of oxidants in NF-κB activation and TNF-α gene
transcription induced by hypoxia and endotoxin. J Immunol.
165:1013–1021. 2000.
|
|
22
|
Lluis JM, Buricchi F, Chiarugi P, Morales
A and Fernandez- Checa JC: Dual role of mitochondrial reactive
oxygen species in hypoxia signaling: activation of nuclear
factor-κB via c-SRC and oxidant-dependent cell death. Cancer Res.
67:7368–7377. 2007.
|
|
23
|
Yokoi K and Fidler IJ: Hypoxia increases
resistance of human pancreatic cancer cells to apoptosis induced by
gemcitabine. Clin Cancer Res. 10:2299–2306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rius J, Guma M, Schachtrup C, Akassoglou
K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG and Karin M:
NF-κB links innate immunity to the hypoxic response through
transcriptional regulation of HIF-1α. Nature. 453:807–811.
2008.
|
|
25
|
Belaiba RS, Bonello S, Zähringer C,
Schmidt S, Hess J, Kietzmann T and Görlach A: Hypoxia up-regulates
hypoxia-inducible factor-1α transcription by involving
phosphatidylinositol 3-kinase and nuclear factor κB in pulmonary
artery smooth muscle cells. Mol Biol Cell. 18:4691–4697. 2007.
|
|
26
|
Huber MA, Azoitei N, Baumann B, Grünert S,
Sommer A, Pehamberger H, Kraut N, Beug H and Wirth T: NF-κB is
essential for epithelial-mesenchymal transition and metastasis in a
model of breast cancer progression. J Clin Invest. 114:569–581.
2004.
|
|
27
|
Shin SR, Sánchez-Velar N, Sherr DH and
Sonenshein GE: 7,12-Dimethylbenz(a)anthracene treatment of a
c-rel mouse mammary tumor cell line induces epithelial to
mesenchymal transition via activation of nuclear factor-κB. Cancer
Res. 66:2570–2575. 2006.PubMed/NCBI
|
|
28
|
Chua HL, Bhat-Nakshatri P, Clare SE,
Morimiya A, Badve S and Nakshatri H: NF-κB represses E-cadherin
expression and enhances epithelial to mesenchymal transition of
mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2.
Oncogene. 26:711–724. 2007.
|
|
29
|
Julien S, Puig I, Caretti E, Bonaventure
J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A
and Larue L: Activation of NF-κB by Akt upregulates Snail
expression and induces epithelium mesenchyme transition. Oncogene.
26:7445–7456. 2007.
|
|
30
|
Wang SJ, Gao Y, Chen H, Kong R, Jiang HC,
Pan SH, Xue DB, Bai XW and Sun B: Dihydroartemisinin inactivates
NF-κB and potentiates the anti-tumor effect of gemcitabine on
pancreatic cancer both in vitro and in vivo. Cancer Lett.
293:99–108. 2010.PubMed/NCBI
|
|
31
|
Kong R, Sun B, Jiang H, Pan S, Chen H,
Wang S, Krissansen GW and Sun X: Downregulation of nuclear
factor-kappaB p65 subunit by small interfering RNA synergizes with
gemcitabine to inhibit the growth of pancreatic cancer. Cancer
Lett. 291:90–98. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Franovic A, Gunaratnam L, Smith K, Robert
I, Patten D and Lee S: Translational up-regulation of the EGFR by
tumor hypoxia provides a non mutational explanation for its
overexpression in human cancer. Proc Natl Acad Sci USA.
104:13092–13097. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang
SY, Liu CJ, Teng SC and Wu KJ: Direct regulation of TWIST by HIF-1α
promotes metastasis. Nat Cell Biol. 10:295–305. 2008.
|
|
34
|
Hotz B, Arndt M, Dullat S, Bhargava S,
Buhr HJ and Hotz HG: Epithelial to mesenchymal transition:
expression of the regulators snail, slug, and twist in pancreatic
cancer. Clin Cancer Res. 13:4769–4776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang B, Zhang Z and Ke Y: Conversion of
cadherin isoforms in cultured human gastric carcinoma cells. World
J Gastroenterol. 12:966–970. 2006.PubMed/NCBI
|
|
36
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nature Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nieto MA: The snail superfamily of
zinc-finger transcription factors. Nat Rev Mol Cell Biol.
3:155–166. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: an alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Alexander NR, Tran NL, Rekapally H,
Summers CE, Glackin C and Heimark RL: N-cadherin gene expression in
prostate carcinoma is modulated by integrin-dependent nuclear
translocation of Twist1. Cancer Res. 66:3365–3369. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Walmsley SR, Print C, Farahi N,
Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM,
Cowburn AS, Johnson N and Chilvers ER: Hypoxia-induced neutrophil
survival is mediated by HIF-1α-dependent NF-κB activity. J Exp Med.
201:105–115. 2005.
|
|
41
|
Carbia-Nagashima A, Gerez J, Perez-Castro
C, Paez-Pereda M, Silberstein S, Stalla GK, Holsboer F and Arzt E:
RSUME, a small RWD-containing protein, enhances SUMO conjugation
and stabilizes HIF-1α during hypoxia. Cell. 131:309–323.
2007.PubMed/NCBI
|