Introduction
As a major public health issue worldwide, colorectal
cancer (CRC) is the third most common type of cancers and the
second leading cause of death by cancer in the Western world
(1). Development of a malignant
colorectal tumor is a progressive process with a duration of
several years. A series of molecular events and alterations, which
involve adhesion molecules, angiogenic factors, chemotactic and
growth factors, appear to be responsible for the different stages
with invasion and metastatic spread (2). However, the predominant drivers
contributing to CRC cell malignancy, such as their migration and
invasion to the liver, remain to be determined.
Inhibitors of DNA-binding proteins (ID1, ID2, ID3
and ID4) are characterized structurally as classic basic
helix-loop-helix (bHLH) transcription factors but lacking a
DNA-binding domain (3). The
functional activation of ID proteins is initiated by forming a
heterodimer with bHLH transcription factors and then blocking their
DNA-binding domain, and as a result, inhibiting transcriptional
activity. It is widely accepted that the accumulation of activated
ID proteins function as dominant-negative regulators of bHLH
transcription factors and are critical for various cellular
processes in mammalian cells. ID1 was identified as a modulator of
E2A to inhibit the cell cycle (4).
Some ID proteins have been shown to neutralize pRB suppression on
E2F-DP1 activity to potentiate S phase progression (5,6) and to
inhibit cell differentiation in cells (7). Consistent with their functional
features, ID genes are expressed abundantly in many proliferating
tissues but scarcely in terminally differentiated tissues (8), suggesting their specific role in
embryonic development (9).
Along with the important cellular function,
dysregulation of ID expression strongly correlates with cancer
progression (10,11). Overexpression of IDs is a
gene-expression signature in a wide variety of cancers. Therefore,
IDs have been recognized as oncogenic or ontogenesis-related
factors. ID1 is one of the most extensively investigated members of
the ID family. Accumulating evidence has confirmed that ID1 is
upregulated in several types of tumors including breast, prostate,
lung, gastric, esophageal and colorectal adenocarcinoma (12,13).
This increase in ID1 expression appears to be crucial for growth
grades, invasive properties and subsequently a poor clinical
outcome (13–18). Evidence also indicates that
expression of ID proteins, including ID1, correlates with the p53
level and the mitotic index in colorectal tumors (19,20).
However, our knowledge concerning the potential roles of ID1 in
proliferation, migration and metastasis of colon cancer is limited
(20).
In the present study, we knocked down ID1 using
lentiviral shRNA to investigate the role of ID1 in CRC cell lines.
We investigated whether depletion of ID1 in CRC cells is associated
with inhibition of proliferation, migration/invasion and distal
metastasis abilities. We also provided evidence that the function
of ID1 in regulating colon cancer cell migration was partly through
the chemokine receptor CXC chemokine receptor 4 (CXCR4) in the
HCT116 cell line.
Materials and methods
Cell culture
Human colon cancer cell lines HCT116 and SW620 were
obtained from the Shanghai Cell Bank of the Chinese Academy of
Sciences. HCT116 cells were cultured in McCoy’s 5A medium, and
SW620 were maintained in L-15 medium supplemented with 10% fetal
bovine serum (FBS). Cells were collected when at least 80%
confluent for the experiments.
Gene knockdown (KD) and
overexpression
All of the shId1 and control plasmids were purchased
from Sigma-Aldrich Corp. The human short hairpins used to target
ID1 were as follows: 5′-AGTCTCTGGTGACTAGTAG-3′ (shID1-1);
5′-TGAGGCGTGAGTAACAGCC-3′ (shID1-2); 5′-ACCTGCTGCTCGTCCAGCA-3′
(shID1-3) and 5′-CATGTCGTAGAGCAGCACG-3′ (shID1-4). A control shRNA
unrelated to the human sequences was used as a negative control.
The shRNA vector was co-transfected with packaging vectors
pCMV-Dr82 and pCMV-VSVG at a ratio of 4:3:2 into 293T cells using
Lipofectamine 2000 reagent (Invitrogen, USA). Polybrene (6 μg/ml;
Sigma, USA) was added to the media for viral infection. For
generating stable clones, the ID1-KD cells and control vector cells
were selected using 1.5 μg/ml puromycin (Merck, Germany) for 3
weeks. The expression of ID1 was routinely detected by western
blotting and quantitative real-time RT-PCR. For CXCR4
overexpression, full-length CXCR4 cDNA was cloned into a pcDNA3.0
vector using standard protocols. ID1 stable KD HCT116 cells were
transfected with either a pcDNA-CXCR4 or a pcDNA3.0 empty vector
using Lipofectamine 2000 reagent.
Flow cytometric analysis
Apoptosis/necrosis was determined using the PE
Annexin V Apoptosis Detection Kit I (BD Biosciences), according to
the manufacturer’s recommendations. Samples were analyzed by flow
cytometry (FACSCalibur; BD Biosciences). The experiments were
performed in triplicate and repeated six times.
Proliferation assay
A total of 4×103 HCT116 or
5×104 SW620 cells/well with ID1-KD or control shRNA were
seeded in 96-well plates and cultured for 48 h. Cell proliferation
was determined using the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt (MTS) assay (Promega, Madison, WI, USA) according to the
manufacturer’s recommendations.
Wound healing assay and
migration/invasion assays
Cells with ID1-KD shRNA or cells with control shRNA
were plated in 6-well plates in duplicate at ~80% confluency and
allowed to grow overnight. The following day a scratch wound was
made through the center of each well using a 10-μl pipette tip.
Plates were washed three times with phosphate-buffered saline
(PBS), and fresh media were then added to remove any loose cells.
After 48 h, the cells were examined by light microscopy to
determine resealing of the monolayer.
A Transwell migration assay was performed using
8.0-μm pore insert 24-well plates (Becton-Dickinson AG, Allschwil,
Switzerland). Transwell chambers were pre-coated with 1 μg/ml
fibronectin on the underside of the membrane. A total of
1×105 HCT116 or 5×105 SW620 cells were plated
in a 24-well cell culture insert in 100 μl of FBS-free media.
Inserts were then placed in the well with 500 μl of 20% FBS
containing media. After 24 h (for HCT116 cells) or 48 h (for SW620
cells), medium and cells in the culture insert were removed. Cells
at the bottom side of the insert were methanol-fixed and stained
with 0.1% crystal violet. Five random fields were selected, and
cells were counted at a ×100 magnification to determine the average
number of cells in each insert. The invasion assay was carried out
in the same manner except that the 8.0-μm pore size membrane insert
was coated with Matrigel (BD Biosciences) that had been diluted in
medium (1:5 dilution).
RNA extraction and real-time reverse
transcription-PCR
Total RNA from the CRC cells was extracted using the
RNeasy Mini kit, according to the manufacturer’s protocol (Qiagen
Inc., USA). Total cellular RNA was isolated from the xenografted
tumors using TRIzol reagent (Invitrogen). One microgram of total
RNA was reverse-transcribed using the Promega Reverse Transcription
System A3500 (Promega). Quantitative real-time polymerase chain
reaction (qRT-PCR) was run on a LightCycler Roche 480 with DyNAmo
Flash SYBR-Green qPCR kit (Thermo Fisher Scientific, USA). The
thermocycling program was performed according to the instrument’s
manual. Primers for the genes of interest are listed in Table I. For the relative quantification of
the mRNA levels, 6 independent amplifications were performed for
each target gene, with triplicate samples. β-actin was used as a
reference gene to normalize gene expression in each sample. The
relative mRNA expression levels were normalized to the level of
β-actin mRNA expression (equal 1) in the corresponding samples. The
data were presented numerically by the comparative
2−ΔΔCt method.
 | Table IPrimers used in the present
study. |
Table I
Primers used in the present
study.
| Gene | | Primer sequence
(5′→3′) |
|---|
| ID1 | Forward |
CGTGCTGCTCTACGACATGA |
| Reverse |
GCTCCAACTGAAGGTCCCTG |
| PCNA | Forward |
AACCTGCAGAGCATGGACTC |
| Reverse |
TCATTGCCGGCGCATTTTAG |
| Survivin | Forward |
TCTCTACATTCAAGAACT |
| Reverse |
TTGAAGCAGAAGAAACAC |
| MMP2 | Forward |
TCTTCCCCTTCACTTTCCTG |
| Reverse |
ACTTGCGGTCATCATCGT |
| MMP9 | Forward |
GCAGAGATGCGTGGAGAGT |
| Reverse |
CCCTCAAAGGTTTGGAATC |
| CXCR4 | Forward |
ATACACTTCAGATAACTAC |
| Reverse |
TAAGAAGATGATGGAGTA |
| β-actin | Forward |
TGGCACCACACCTTCTACA |
| Reverse |
AGCACAGCCTGGATAGCA |
Western blot analysis
Cells in 6-cm dishes were washed with cold PBS and
harvested by scraping following addition of lysis buffer (50 mM
HEPES, pH 7.4, 250 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 1 mM
Na3VO4, 1 mM NaF, 1 mM PMSF, 1 mM
dithiothreitol and a protease inhibitor cocktail from Roche) for 15
min on ice. The protein concentration was determined by the
bicinchoninic acid assay (BCA Protein Assay kit; Pierce, USA). The
samples were boiled for 5 min and stored at -20°C until use. Equal
amounts of protein were electrophoresed on polyacrylamide gradient
gels (10–15%; Bio-Rad Laboratories) and electro-transferred to
membranes. After transfer, the membranes were then blocked in 3%
bovine serum albumin at room temperature for 2 h. The membranes
were then incubated overnight at 4°C with primary antibodies
against PCNA, survivin (Abcam Inc., Cambridge, MA, USA), ID1, MMP2
and MMP9, (Santa Cruz Biotechnology, Santa Cruz, CA, USA), CXCR4
(Cell Signaling Technology, Beverly, MA, USA) and β-actin (Santa
Cruz Biotechnology). The binding of secondary horseradish
peroxidase-conjugated antibodies was visualized by enhanced
chemiluminescence (ECL Plus, USA).
Immunohistochemical staining
Immunohistochemistry for detection of target protein
(ID1, PCNA, survivin, MMP2, MMP9 and CXCR4) expression in the tumor
tissues from mice was carried out using the
streptavidin-biotin-peroxidase (SP) staining method. The
immunostaining SP kit was purchased from Fuzhou Maxim Biotech
Company. Paraffin-embedded sections were deparaffinized by xylene
and dehydrated in graded alcohol. To retrieve antigen, the sections
were boiled in 10 mM citrate buffer (pH 6.0) for 5 min. The tissue
sections were treated with peroxidase blocking agent to block
endogenous peroxidase and normal rabbit serum to block non-specific
binding sites. The primary antibodies were used at a dilution
according to the antibodies manual and were added to adjacent
tissue sections and incubated overnight at 4°C. Secondary
antibodies were added to the sections and incubated at room
temperature for 10 min. S-P complex was added at room temperature
for 10 min, DAB was used for the color reaction, and the sections
were counterstained with hematoxylin. Stained sections were viewed
and photographed using a fluorescence microscope.
Gelatin zymography
Gelatin zymography was carried out on protein
extracts from the HCT116 and SW620 cells. Cells were plated at a
density of 1×106 in 6-well plates. After 24 h, the cells
were washed with PBS and incubated in 1 ml of serum-free medium for
24 h. The conditioned medium was separated on 10% SDS/PAGE with 1
mg/ml gelatin incorporated into the gel mixture. Following
electrophoresis at 4°C, the gels were washed 4 times for 15 min
each in 2.5% Triton X-100 to remove the SDS and were incubated for
37 h at 37°C in 50 mM Tris, pH 7.5, 10 mM CaCl2, 1 μM
ZnCl2 and 150 mM NaCl. Afterwards, the gels were fixed
and stained with 0.5% Coomassie blue in 30% isopropanol/10% acetic
acid for 1 h, then destained in 30% isopropanol/10% acetic acid.
The stain was washed out with water until clear bands were
observed.
In vivo nude mouse study
Male nude mice (BALB/c nu/nu), 4–6 weeks old,
weighing ~16–19 g, were purchased from the Shanghai SLAC Laboratory
Animal Co., Ltd. (Shanghai, China). All experiments were approved
by the Animal Ethics Committee of Fujian Medical University. To
establish CRC xenografts, six mice received 3×106
control and shID1-KD HCT116 cells in a final volume of 0.1 ml PBS
by subcutaneous injection in the right and left groin,
respectively. Measurement of the resulting tumor xenografts began
when the size was >2 mm in diameter and was carried out
thereafter every three days. Tumor volumes were calculated using
the following formula: Volume = [length (mm) × width2
(mm2)]/2. Mice were sacrificed 21 days after the
injections and immediately weighed.
For the experimental liver metastasis assays, cells
were injected into the spleen (5×106 cells/mouse). The
mice were placed in the right lateral decubitus position, an
incision in the abdominal wall on the left side was made, the
spleen was exteriorized, and the cells were injected into the
spleen. Mice were sacrificed at day 28. Tumor samples from the site
of the tumor injection and from livers (metastasis target organ)
were shock-frozen in liquid nitrogen, formalin-fixed and
paraffin-embedded.
Statistical analysis
The data represent means ± SD from at least six
independent experiments. Statistical analysis was performed with
the Student’s t-test at a significance level of P<0.05. All data
analyses were conducted by use of the SPSS 20.0 statistical
software package.
Results
Downregulation of ID1 decreases
proliferation and induces apoptosis
In order to determine the function of ID1 in CRC, we
established stable cancer cell lines with ID1 knockdown. To this
end, 4 independent lentiviral shRNAs targeting the ID1 gene were
evaluated for their knockdown efficiency. Quantitative PCR (qPCR)
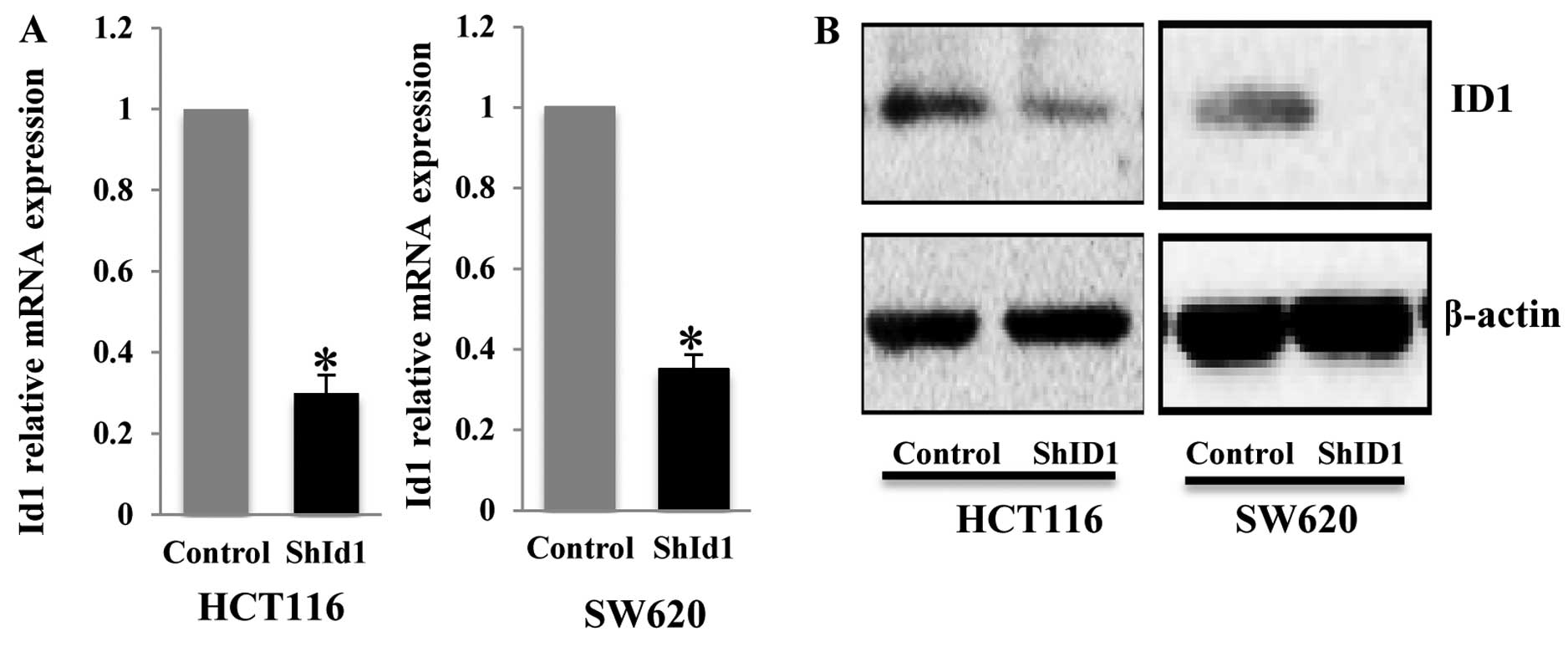
analysis demonstrated that ID1 transcript in the HCT116 cell line
was affected by the different ID1 shRNAs to varying degrees of
reduction (50–85%, data not shown). We used the lentivirus
containing the most efficient shRNA to infect the HCT116 and SW620
cells and found a robust depletion of ID1 mRNA in both cell lines
(Fig. 1A). Subsequently, we
generated stable pools of HCT116 and SW620 cells with shID1 and
control shRNA. Western blot analysis demonstrated an ~60–70%
reduction in ID1 protein in the ID1-KD cells as compared to the
control cells (Fig. 1B).
To assess the function of ID1 in the proliferation
of CRC cells, we performed an MTS assay to determine the cell
proliferation rate in the ID1-KD and control cells. As shown in
Fig. 2A, ID1 knockdown in both
HCT116 and SW620 cells resulted in a significant decrease in cell
proliferation when compared to the controls, suggesting its pivotal
role in cell growth. We then aimed to ascertain whether depletion
of ID1 induces apoptosis in CRC cells. To detect early and late
apoptosis rates in shID1 knockdown CRC cells, the Annexin V-PE and
7-AAD double staining assay was used. The early (Annexin
V+/7AAD−) and late apoptosis (Annexin
V+/7-AAD+) rates were significantly increased
in the ID1-KD cells when compared to these rates in the controls
(Fig. 2B). Two genes associated
with proliferation (PCNA) and survival (survivin) were examined by
real-time PCR and western blotting. Consistent with these
observations, the mRNA quantities of PCNA and survivin were
significantly decreased in the ID1-KD HCT116 and SW620 cells
(P<0.05) (Fig. 2C). Western
blotting also confirmed that protein levels of PCNA and survivin
were reduced in the ID1-KD cells when compared to the levels in the
control cells (Fig. 2D).
Collectively, these data indicate that ID1 plays an important role
in CRC cell proliferation and apoptosis and downregulation of ID1
forms a limiting factor for growth of these cancer cells.
Downregulation of ID1 reduces motility,
migration and invasion capacity of CRC cells
To analyze a possible effect of ID1-KD on the
migration of CRC cells, cell migration ability was assessed with a
scratch assay. As shown in Fig. 3A,
ID1-KD cells migrated through the wound scratch more slowly than
the control cells. Migration/invasion capacity was tested by
Transwell/invasion assays. We also found a significant decrease in
cellular migration/invasion capacity in cells with ID1 knockdown.
The decrease in the migration/invasion of ID1-KD cells was
statistically significant (P<0.05 vs. control group) (Fig. 3B–D).
MMP2 and MMP9 belong to the gelatinase subfamily of
matrix metalloproteinases, which are known to be involved in
invasion of cancer cells. We then tested the expression of these
markers in the control and ID1-KD cells. Both qPCR and western blot
assays revealed that mRNA and protein of MMP2 and MMP9 were
significantly decreased in the ID1-KD HCT116 and SW620 cells
(Fig. 4A and B). Consistently,
gelatin zymography revealed an anticipated decrease in MMP2 and
MMP9 activity in the ID1-KD cells compared with the control cells
(Fig. 4C). In addition, we examined
the alteration in CXCR4 in the ID1-KD cells. CXCR4 is a chemokine
receptor closely linked to cancer cell growth, migration and
invasion in CRC and other cancer types (23). We found that CXCR4 mRNA and protein
levels were concurrently decreased in the HCT116 and SW620 cells
with ID1 knockdown (Fig. 4A and B).
These results revealed a possible molecular mechanism underlying
ID1-mediated cell migration and invasion.
CXCR4 reverses the negative effect of the
downregulation of ID1 in regards to migration and invasion of CRC
cells
Our data suggest that the attenuation of motility,
migration and invasion capacity of CRC cells by ID1 depletion are
attributable to the compromised function of MMP proteins and CXCR4.
To substantiate this hypothesis, CXCR4, as a representative, was
further investigated by using a genetic rescue experiment, in which
CXCR4 was compensated by its exogenous protein in ID1-KD HCT116
cells. As shown in Fig. 5A, CXCR4
in the ID1-KD cells was restored to a similar level as that in the
controls by transfection of a CXCR4-overexpression vector.
Consequently, ID1-KD/CXCR4-overexpressing (OE) cells showed higher
motility in the scratch assay than the ID1-KD counterparts
(Fig. 5B). Moreover, in the
Transwell culture assays, CXCR4 restoration significantly improved
migration (Fig. 5C and D) and
invasion of ID1-KD cells (Fig. 5E).
Although CXCR4 rescue did not fully reverse the effect of ID1
knockdown, this partial rescue function in regards to the migration
and invasion capacity indicated that CXCR4 indeed played an
important role in the altered tumor cell motility by ID1 knockdown.
This finding also suggests that other factors are likely to
orchestrate this process.
Downregulation of ID1 inhibits tumor
growth of HCT116 cells in a xenograft mouse model in vivo
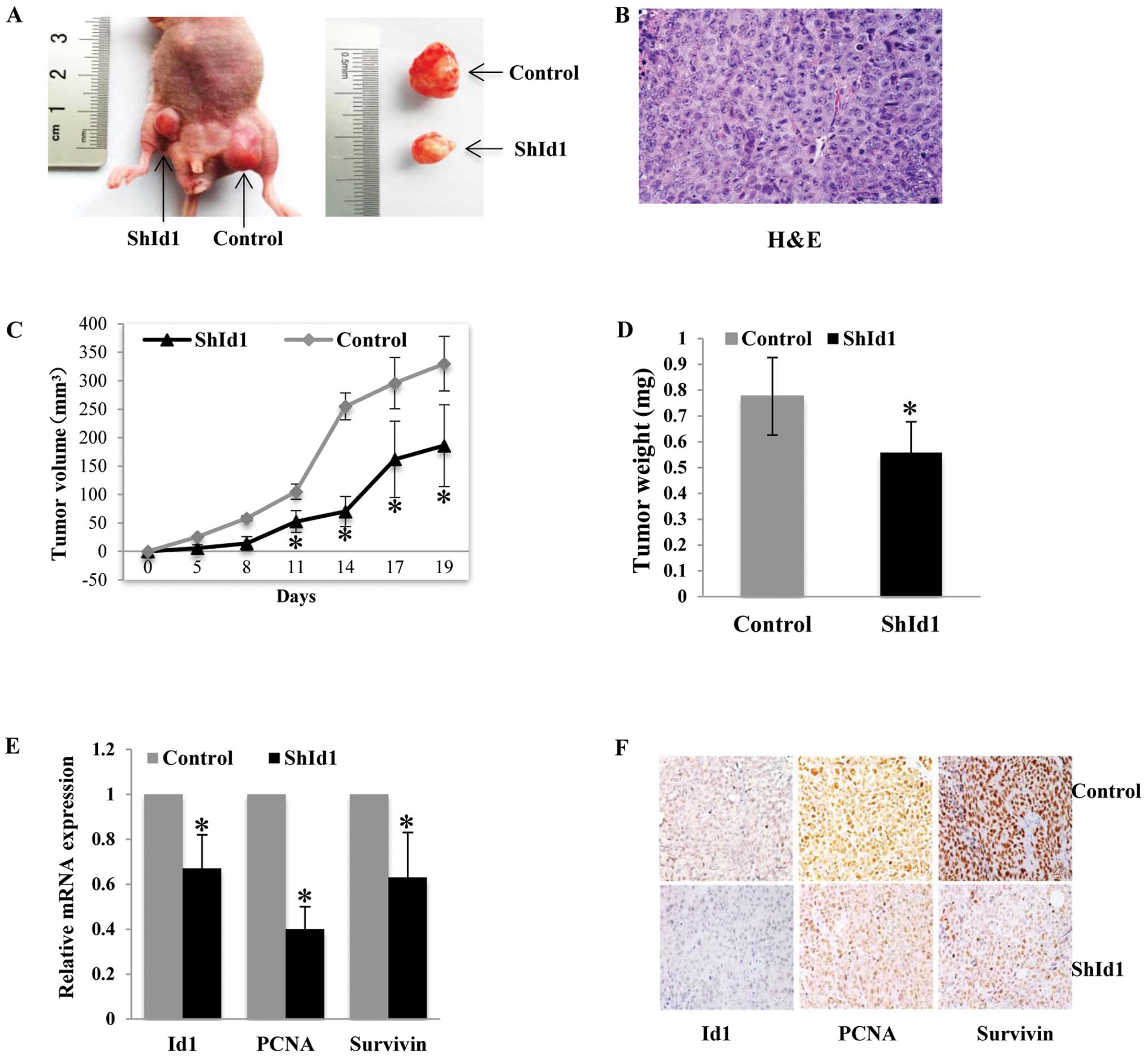
The effects of ID1 knockdown on tumor growth in
vivo were first investigated in a subcutaneous tumor model
using HCT116 cells. In agreement with the in vitro findings,
ID1-KD cells formed smaller tumors than the control cells (Fig. 6A–D). The effect of shId1 on the
silencing of the Id1 gene in the xenografted tumors was evaluated
by real-time PCR and immunohistochemical analysis. As shown in
Fig. 6E and F, a >50% ID1 mRNA
and protein reduction was found in the ID1-KD tumors compared with
the controls (P<0.05). In addition, PCNA and survivin mRNA
levels were significantly decreased in the xenografted tumors
(P<0.05). The protein levels of PCNA and survivin assayed by
immunohistochemistry were also decreased in the ID1-KD tumor
sections (Fig. 6F).
Downregulation of ID1 inhibits metastasis
of HCT116 cells in a liver metastasis mouse model
Next, we evaluated the effect of ID1 on the in
vivo liver metastasis of HCT116 KD tumors. In line with our
in vitro findings, both qPCR and immunohistochemistry showed
decreased MMP2, MMP9 and CXCR4 mRNA and protein in the ID1-KD
subcutaneous tumors compared to the control group (Fig. 7A and B). We implanted HCT116 cells
into the spleen which led to tumor growth in the spleen and
metastasis to the liver. Twenty-eight days after tumor cell
injection, all animals in the control group developed liver
metastases (10/10). In contrast, only 6/10 animals in the ID1-KD
group developed liver metastases that were barely visible
(P<0.05). As compared to the vector control, ID1 profoundly
suppressed liver metastases in the mice (Fig. 7C and D).
Discussion
The principal finding in the present study was that
knockdown of ID1 protein expression correlates with the growth
arrest of CRC cell lines and the suppression of hepatic metastasis
of CRC tumors in a mouse model. The study also emphasized the role
of ID1/CXCR4 as a positively regulatory axis required for CRC cell
proliferation and migration and tumor invasion. This finding is
consistent with these of recent reports on the inhibitive effect of
ID1 and ID3 gene downregulation on CRC hematogenous metastasis at
the early stage of the tumor (21).
The results imply that the ID1 gene could be a therapeutic target
for CRC. Our present study indicated that ID1 knockdown caused
reduced proliferation and induced apoptosis of CRC cells in
vitro and restrained tumor growth in vivo. This can be
interpreted along with recent studies of the tumorigenic effect of
ID1 in keratinocytes (22) and in a
variety of human tumors (23,24),
which suggest that an elevated ID1 protein level may promote cell
proliferation, inhibit cellular apoptosis, and repress
differentiation, thereby leading to the onset and progression of
CRC.
Since survivin was reported to be a downstream
regulator in the ID1/PI3K/Akt/NF-κB/survivin signaling pathway for
endothelial progenitor cell proliferation, and CRC tumor growth and
malignant metastasis (25,26), the fact that ID1 knockdown induced a
decrease in survivin expression, may indicate that activation of
survivin is a consequence of the expression of ID1 in CRC cell
lines. It has been suggested that PCNA is a proliferation marker of
tumors and is critical for DNA synthesis (27). We showed that ID1 knockdown
decreased PCNA expression at both the mRNA and protein levels.
However, no significant change in the cell cycle was noted
following ID1 knockdown (data not shown). Thus, there may be
alternative mechanisms that remain to be identified in the
stabilization of DNA synthesis. To some extent, these results,
together with previous studies, imply that modulation of ID1
expression positively interferes with the functions of PCNA and
survivin and subsequently inhibits tumor growth and invasion in
CRC.
Many reports have addressed the role of ID1 in tumor
metastasis. For example, ID1 was found to elevate the expression of
epithelial mesenchymal markers in nicotine and EGF-induced
proliferation, migration and invasion of non-small cell lung cancer
(23). Similarly, ID1 was found to
be positively associated with the migratory and invasive features
of breast cancer cells by enhancing epithelial-mesenchymal-like
markers (28). A clinical
observation indicated that ID1 expression was strongly associated
with lymph node metastasis in patients with CRC (13). Our results support recent study by
O’Brien et al (20) who
demonstrated a significant reduction in tumor growth and hepatic
metastatic burden following double-gene ID1 and ID3 knockdown in
CRC cell lines in a mouse model. We also found that ID1 knockdown
suppressed the expression and activity of the matrix
metalloproteinases, MMP2 and MMP9, in vitro, suggesting that
ID1-KD may protect against angiogenic destruction of the
extracellular matrix of regional blood vessels during adjacent and
distal invasion and metastasis (29–31).
ID1 knockdown induced suppression of angiogenesis may likely be
explained in part by the ID1/NF-κB/MMP-2 or ID1/PI3K/Akt signaling
pathways identified in ovarian cancer (32) and integrins (α3, α6 and β1)/laminin
adhesion in pancreatic cancer (33).
Another finding of the present study indicated that
gross overexpression of CXCR4 reversed the ID1
knockdown-suppressive effects on cell proliferation and metastasis.
Metastasis-related molecular events have been explored in several
studies (34–36) of which one of the chemokine family
members, CXCR4, was significantly higher in liver metastasis
compared with primary CRC tumor tissue and was found to correlate
with reduced overall median patient survival after liver metastasis
(34). In addition, a specific
ligand of CXCR4, stromal cell-derived factor-1 (SDF-1), was found
to highly express in tumor-adherent stromal cells of metastatic
organs such as the liver, lung and lymph nodes (36). Notably, we demonstrated both in
vitro and in vivo, significant reductions in CXCR4
expression and hepatic metastasis burden of CRC cells following
inactivation of ID1. The fact that overexpression of CXCR4 largely
restored the migration and invasion capacities of the HCT116 ID1
knockdown cells, probably represents a positive loop of feedback
underlying the interaction of ID1 and CXCR4 in CRC migration and
invasion ability. Taken together, the present data suggest that
highly enforced expression of ID1 causes CRC cell growth arrest and
retards metastatic progression in a CXCR4-dependent manner.
In summary, our studies in vitro and in
vivo confirmed the regulatory role of ID1 in the proliferation
and metastasis of CRC. The findings provide initial evidence that
downregulation of ID1 reduces CRC migration and invasion partly
through reduced CXCR4 expression in tumor cells, suggesting the
rationale of the ID1 protein as a clinical target of CRC treatment.
In order to elucidate its oncogenic function, additional studies
are needed to explore the network of ID1 within CXCR4/CD133-related
stromal self-renewal in CRC malignancy (37).
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (grant no. 81250002).
We would like to thank Professor Xin Lin for his expert suggestions
in experiments.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2
|
Christofori G: New signals from the
invasive front. Nature. 441:444–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Benezra R, Davis RL, Lockshon D, et al:
The protein Id: a negative regulator of helix-loop-helix DNA
binding proteins. Cell. 61:49–59. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Prabhu S, Ignatova A, Park ST and Sun XH:
Regulation of the expression of cyclin-dependent kinase inhibitor
p21 by E2A and Id proteins. Mol Cell Biol. 17:5888–5896.
1997.PubMed/NCBI
|
|
5
|
Iavarone A, Garg P, Lasorella A, et al:
The helix-loop-helix protein Id-2 enhances cell proliferation and
binds to the retinoblastoma protein. Genes Dev. 8:1270–1284. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lasorella A, Iavarone A and Israel MA: Id2
specifically alters regulation of the cell cycle by tumor
suppressor proteins. Mol Cell Biol. 16:2570–2578. 1996.PubMed/NCBI
|
|
7
|
Lasorella A, Uo T and Iavarone A: Id
proteins at the cross-road of development and cancer. Oncogene.
20:8326–8333. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yokota Y and Mori S: Role of Id family
proteins in growth control. J Cell Physiol. 190:21–28. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lyden D, Young AZ, Zagzag D, et al: Id1
and Id3 are required for neurogenesis, angiogenesis and
vascularization of tumour xenografts. Nature. 401:670–677. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Norton JD: ID helix-loop-helix proteins in
cell growth, differentiation and tumorigenesis. J Cell Sci.
113:3897–3905. 2000.PubMed/NCBI
|
|
11
|
Sikder HA, Devlin MK, Dunlap S, et al: Id
proteins in cell growth and tumorigenesis. Cancer Cell. 3:525–530.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang HY, Liu HL, Liu GY, et al: Expression
and prognostic values of Id-1 and Id-3 in gastric adenocarcinoma. J
Surg Res. 167:258–266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao ZR, Zhang ZY, Zhang H, et al:
Overexpression of Id-1 protein is a marker in colorectal cancer
progression. Oncol Rep. 19:419–424. 2008.PubMed/NCBI
|
|
14
|
Forootan SS, Wong YC, Dodson A, et al:
Increased Id-1 expression is significantly associated with poor
survival of patients with prostate cancer. Hum Pathol.
38:1321–1329. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luo KJ, Wen J, Xie X, et al: Prognostic
relevance of Id-1 expression in patients with resectable esophageal
squamous cell carcinoma. Ann Thorac Surg. 93:1682–1688. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun W, Guo MM, Han P, et al: Id-1 and the
p65 subunit of NF-κB promote migration of nasopharyngeal carcinoma
cells and are correlated with poor prognosis. Carcinogenesis.
33:810–817. 2012.
|
|
17
|
Cheng YJ, Tsai JW, Hsieh KC, et al: Id1
promotes lung cancer cell proliferation and tumor growth through
Akt-related pathway. Cancer Lett. 307:191–199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sumida T, Murase R, Onishi-Ishikawa A, et
al: Targeting Id1 reduces proliferation and invasion in aggressive
human salivary gland cancer cells. BMC Cancer. 13:1412013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wilson JW, Deed RW, Inoue T, et al:
Expression of Id helix-loop-helix proteins in colorectal
adenocarcinoma correlates with p53 expression and mitotic index.
Cancer Res. 61:8803–8810. 2001.PubMed/NCBI
|
|
20
|
O’Brien CA, Kreso A, Ryan P, et al: ID1
and ID3 regulate the self-renewal capacity of human colon
cancer-initiating cells through p21. Cancer Cell. 21:777–792.
2012.PubMed/NCBI
|
|
21
|
Okaji Y, Tsuno NH, Kitayama J, et al:
Effects of down-regulating the Id genes in human colorectal
cancer cells on early steps of haematogenous metastasis. Eur J
Cancer. 42:668–673. 2006.PubMed/NCBI
|
|
22
|
Alani RM, Young AZ and Shifflett CB: Id1
regulation of cellular senescence through transcriptional
repression of p16/Ink4a. Proc Natl Acad Sci USA. 98:7812–7816.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pillai S, Rizwani W, Li X, et al: ID1
facilitates the growth and metastasis of non-small cell lung cancer
in response to nicotinic acetylcholine receptor and epidermal
growth factor receptor signaling. Mol Cell Biol. 31:3052–3067.
2011. View Article : Google Scholar
|
|
24
|
Li W, Zhang CH, Hong YL, et al: Inhibitor
of DNA-binding-1/inhibitor of differentiation-1 (ID-1) is
implicated in various aspects of gastric cancer cell biology. Mol
Biol Rep. 39:3009–3015. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li W, Wang H, Kuang CY, et al: An
essential role for the Id1/PI3K/Akt/NFκB/survivin signalling
pathway in promoting the proliferation of endothelial progenitor
cells in vitro. Mol Cell Biochem. 363:135–145. 2012.PubMed/NCBI
|
|
26
|
Ye Q, Cai W, Zheng Y, et al: ERK and AKT
signaling cooperate to translationally regulate survivin expression
for metastatic progression of colorectal cancer. Oncogene. Apr
29–2013.(Epub ahead of print). View Article : Google Scholar
|
|
27
|
Zhong W, Peng J, He H, et al: Ki-67 and
PCNA expression in prostate cancer and benign prostatic
hyperplasia. Clin Invest Med. 31:E8–E15. 2008.PubMed/NCBI
|
|
28
|
Tobin NP, Sims AH, Lundgren KL, Lehn S and
Landberg G: Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer.
11:4172011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li B, Tsao SW, Li YY, et al: Id-1 promotes
tumorigenicity and metastasis of human esophageal cancer cells
through activation of PI3K/AKT signaling pathway. Int J Cancer.
125:2576–2585. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ling YX, Tao J, Fang SF, Hui Z and Fang
QR: Downregulation of Id1 by small interfering RNA in prostate
cancer PC3 cells in vivo and in vitro. Eur J Cancer Prev. 20:9–17.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sato Y: Molecular diagnosis of tumor
angiogenesis and anti-angiogenic cancer therapy. Int J Clin Oncol.
8:200–206. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Su Y, Gao L, Teng L, Wang Y, et al: Id1
enhances human ovarian cancer endothelial progenitor cell
angiogenesis via PI3K/Akt and NF-κB/MMP-2 signaling pathways. J
Transl Med. 11:1322013.PubMed/NCBI
|
|
33
|
Shuno Y, Tsuno NH, Okaji Y, et al: Id1/Id3
knockdown inhibits metastatic potential of pancreatic cancer. J
Surg Res. 161:76–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim J, Takeuchi H, Lam ST, et al:
Chemokine receptor CXCR4 expression in colorectal cancer
patients increases the risk for recurrence and for poor survival. J
Clin Oncol. 23:2744–2753. 2005.
|
|
35
|
Koizumi K, Hojo S, Akashi T, Yasumoto K
and Saiki I: Chemokine receptors in cancer metastasis and cancer
cell-derived chemokines in host immune response. Cancer Sci.
98:1652–1658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kucia M, Reca R, Miekus K, et al:
Trafficking of normal stem cells and metastasis of cancer stem
cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4
axis. Stem Cells. 23:879–894. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang SS, Han ZP, Jing YY, et al:
CD133+CXCR4+ colon cancer cells exhibit
metastatic potential and predict poor prognosis of patients. BMC
Med. 10:852012.PubMed/NCBI
|