Introduction
Glioblastoma multiforme (GBM) is the most malignant
and aggressive tumor, and GBM patients have an extremely poor
prognosis and a mean survival time of less than 2 years, even
following treatment with recent concomitant chemoradiation
(1,2).
Temozolomide (TMZ) is an alkylating agent that
induces DNA methylation of guanine at the O6
position and triggers mismatch repair, which leads to arrest of the
cell cycle and apoptosis (3).
O6-methylguanine-DNA methyltransferase (MGMT) is
well known for removing methyl groups from the O6
position of guanine and contributing to TMZ resistance induction
(4). Several clinical studies have
demonstrated that high MGMT expression through the methylation of
the MGMT promoter is one of the genuine mechanisms responsible for
TMZ resistance. Thus, novel therapeutics that aim to suppress TMZ
resistance by deleting MGMT have been pursued, and
O6-benzylguanine has been considered to be a
promising therapeutic candidate. However, clinical trials did not
show the restoration of TMZ resistance (5,6).
Considering that no tools presently exist to treat TMZ-resistant
(TMZ-R) recurrent glioblastoma, novel therapeutic approaches that
regulate MGMT expression and restore the sensitivity to TMZ are
highly required.
Moreover, it has been suggested based on multi-omic
analysis that mechanisms other than MGMT may trigger TMZ
resistance. Several novel biomarkers that are linked to MGMT
expression and methylation status, such as the HOX signature and
EGFR expression (7), somatic
mutations of the mismatch repair gene MSH6 (8), prolyl 4-hydroxylase, β polypeptide
(P4HB) (9), mutated EGFR (EGFRvIII)
(10) and CD74 (11), have been reported. Regarding novel
approaches to overcome MGMT-related resistance to TMZ, bortezomib
as a proteasome inhibitor (12),
telomerase inhibition (13), a
combination of interleukin (IL)-24 with TMZ (14) and inactivation of MGMT by gene
therapy (15) have been
demonstrated to show moderate effects on MGMT downregulation and
tumor cell death.
According to a recent molecular classification study
of various glioblastomas, it is known that a mesenchymal signature
expressing STAT3 and the C/EBPβ genes is closely linked to poor
prognosis and TMZ resistance with tumor recurrence (16–19).
These genes could be new possible targets for overcoming TMZ
resistance in GB. In contrast, MGMT is not classified into any
specific subtype marker, including the mesenchymal signature
(17). In the present study, we
identified the YKL-40 and MAGEC1 genes as TMZ resistance-associated
biomarkers in the TMZ-R U87 cell line, which showed an obvious
resistance in vivo. Furthermore, we focused on the YKL-40
and STAT3 mechanisms and proposed a restoration model of TMZ
resistance.
Materials and methods
Cell lines
The human glioblastoma cell lines LN18, T98G and U87
were purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA). All cell lines were cultured in Dulbecco’s
modified Eagle’s medium (DMEM) (Sigma, St. Louis, MO, USA)
supplemented with 10% fetal bovine serum (FBS; Invitrogen),
penicillin and streptomycin.
Antibodies and reagents
Antibodies against STAT3, phospho-specific STAT3
(Tyr705), MGMT and β-actin were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA) and Becton-Dickinson (BD)
Biosciences (Franklin Lakes, NJ, USA) for western blotting (WB). A
mouse anti-human YKL-40 monoclonal antibody (MoAb) was purchased
from Abcam (Cambridge, MA, USA) for immunohistochemical (IHC)
studies. Short hairpin (sh)RNAs specific for the human STAT3 or
YKL-40 genes were purchased from Qiagen GmbH (Hilden, Germany). TMZ
was purchased from Sigma-Aldrich (St. Louis, MO, USA) and was
suspended in sterile 0.5% w/v methyl cellulose 400cp solution
(Wako, Tokyo, Japan).
Establishment of the TMZ-resistant U87
cell line
The U87 parental cell line, which is sensitive to
TMZ, was first maintained in low doses of TMZ (5 μM) and then
successively exposed to incremental doses of TMZ (up to 150 μM).
After the killing of a majority of the cells, the surviving cells
were maintained until a normal rate of growth was obtained. The
IC50 value of TMZ was evaluated using the WST-1
assay.
Cell proliferation assay
Cell proliferation was examined using the WST-1
assay (Dojindo Laboratories, Kumamoto, Japan) as described
previously (20). Briefly,
1–2×104 human glioma cells were seeded into each well of
a 96-well microculture plate (Corning, NY, USA). After 4 days, the
WST-1 substrate was added to the culture and the optical density
(OD) was measured at 450 and 620 nm using an immunoreader (Immuno
Mini NJ-2300, Nalge Nunc International, Roskilde, Denmark). The
IC50 value was defined as the dose required for a 50%
reduction in the OD calculated from the survival curve. Percent
survival was calculated as follows: (mean OD of test wells - mean
OD of background wells)/(mean OD of control wells - mean OD of
background wells).
DNA microarray analysis
Total RNA from the U87 parental and TMZ-R cell lines
was extracted using the NucleoSpin RNA II kit (Takara Bio Inc.,
Shiga, Japan) according to the manufacturer’s instructions. One
microgram of total RNA, which was qualified using an Agilent 2100
Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), was
amplified to 100 μg of cRNA and hybridized to a high-density
oligonucleotide array (GeneChip Human Genome U133 Plus 2.0 array;
Affymetrix, Santa Clara, CA, USA). The intensity for each feature
of the array was calculated using the GeneSpringGX ver11 (Agilent)
software. To calculate the change in the average intensity,
normalization for all probe sets was performed. Genes whose
expression was significantly altered by >5-fold at the 5th and
20th passages of the TMZ-R U87 cell line compared to the U87
parental cell line were analyzed.
Inhibition of STAT3 or YKL-40 gene
expression using shRNA transfection of the TMZ-resistant U87
cells
shRNA gene transfection into the TMZ-R U87 cell line
was performed using a lipofection FreeStyle MAX reagent (Life
Technologies, Carlsbad, CA, USA). Four micrograms of plasmid at 1
mg/ml containing STAT3 or YKL-40 shRNA (SureSilencing shRNA vector;
Qiagen) and the same dose of FreeStyle MAX reagent were suspended
in 100 μl of Opti-MEM I reduced-serum medium (Life Technologies),
which was then mixed and incubated for 15 min at room temperature
(RT). The solution was added to 2×106 TMZ-R U87 cells
and incubated at 37°C for 1 h. After washing, the cells were
incubated in DMEM + 10% FBS, harvested on day 3 of culture and
utilized for in vitro and in vivo experiments.
Cell invasion assay
Invasion assays using parental U87 and TMZ-R U87
cells were performed using Matrigel-coated (0.33 mg/ml) Transwell
inserts with a 8-μm pore size (BD Biosciences, Franklin Lakes, NJ,
USA). Cells at 1×105/ml (500 μl) were added to
Transwells in triplicate, and 750 μl of DMEM containing 10% FBS was
added to the lower wells. After 12–18 h of incubation, the cells
that invaded through the membrane were fixed and stained with
Diff-Quik II solution (Dade Behring AG, Germany). Migrated cells
were counted using microscopy.
Quantitative polymerase chain reaction
(qPCR) analysis
Real-time PCR analysis of 10 genes that were rated
as significantly changed at the expression level (>10-fold,
P<0.05) in the TMZ-R U87 cells compared to the parental cells
was performed using the 7500 Real Time PCR System (Applied
Biosystems, Foster, CA, USA) as described previously (20). Additionally, other stem cell and
neuronal markers, GB mesenchymal type markers and STAT3 target
genes were analyzed. Briefly, all PCR primers (CD24, YKL-40, GDF15,
HLA-DQA1, MAGEC1, MGMT, MMP1, AMIGO2, NMU and RFC2 for
expression-altered genes; ABCB1, ALDH1A1, CD44, EGFR, ESA, GFAP,
KLF4, NANOG, NES, OLIG2, Oct3/4, CD133, SOX2, TGFBR2, TUBB3 and VIM
for GB stem cell markers; CDH2, CDH11, COL1A2, FN1, FOXC2, MMP2,
MMP3, SNAIL1, SNAIL2, TCF4, TWIST1, WNT5A, WNT5B, KRT19, CTNNB1,
GSK3B, NOTCH1, PTK2, SIP1, SMAD2 and ZEB1 for EMT-associated genes;
STAT3, C/EBP, bHLH-B2, RUNX1, FOSL2 and ZNF238 for GB mesenchymal
type markers; and BCL2, Bcl-XL, Survivin, cyclin D1, c-Myc, CXCL10,
VEGFR2, MMP9, TGFB1, P53, VEGFA, VEGFC and HIF-1α for STAT3 target
genes) and TaqMan probes were purchased from Applied Biosystems.
Total RNA was extracted from parental U87 and TMZ-R U87 cells.
Complementary DNA was synthesized from 100 ng of total RNA, and
qPCR was carried using a TaqMan RNA-to-Ct 1-Step kit (Applied
Biosystems).
Western blotting (WB)
TMZ-R U87 cells transfected with or without STAT3
and YKL-40 shRNAs were lysed using RIPA buffer (Thermo Fisher
Scientific Inc., Rockford, IL, USA) containing protease and
phosphatase inhibitors and used for WB as described previously
(21). Briefly, the cell lysates
were subjected to SDS-PAGE with a 7.5% polyacrylamide separating
gel and then transferred to PVDF membranes. After blocking, the
membranes were incubated at 4°C overnight with a primary antibody
against STAT3, phospho-specific STAT3, MGMT, YKL-40 or β-actin
(1:200–1:2,000) in blocking solution. After washing, the membranes
were incubated for 1 h with horseradish peroxidase (HRP)-conjugated
anti-mouse IgG (1:5,000). Membranes were treated with ECL Plus
reagent (GE Healthcare) and analyzed using a chemiluminescence
scanner (LAS-3000; Fujifilm, Tokyo, Japan).
ELISA for human YKL-40
YKL-40 levels in the supernatant of parental U87 or
TMZ-R U87 cells were measured using human YKL-40-specific ELISA.
Cells were suspended in DMEM + 2% FBS medium and plated in 96-well
microplates (Corning Inc., Corning, NY, USA) at 4×104
cells (200 μl cells at 2×105/ml)/well. The supernatants
at 24, 48, 72 and 96 h of the culture were collected and YKL-40
levels were measured.
Animal experiments
Male nude mice (BALB/cA-nu/nu, 5–6 weeks of
age) were obtained from Nippon Clea (Tokyo, Japan), housed in a
separate experimental room and given sterilized food and water
ad libitum. All animals were cared for and used humanely
according to Guidelines for the Welfare and Use of Animals in
Cancer Research (22), and all
procedures were approved by the Animal Care and Use Committee of
Shizuoka Cancer Center Research Institute.
U87 (1×106) and U87/TMZ-R cells
(1×106) were inoculated into the flanks of
BALB/cA-nu/nu mice. To evaluate the antitumor activity
against subcutaneous (s.c.) inoculated tumors, the tumor volume (V)
was calculated based on the National Cancer Institute formula as
follows: V (mm3) = length (mm) × [width
(mm)]2 × 1/2.
TMZ was administered orally daily from day 0 to 4 at
the dose of 5 mg/kg. The efficacy of TMZ against the human tumor
cells that were inoculated into the nude mice was expressed as the
mean V/V0 value, where V is the tumor volume on the day
of evaluation and V0 is that on the day of treatment.
The tumor-control (T/C) value was calculated as the mean
V/V0 value of the treated group vs. that of the
untreated group.
For the in vivo experiments using YKL-40 gene
inhibition, mock or YKL-40 shRNA-4-transfected TMZ-R U87 cells were
harvested 3 days after gene transfection and inoculated into 5 nude
mice per shRNA group.
Statistical analysis
Significant differences were analyzed using the
Student’s t-test. Values of P<0.05 were considered to indicate
statistically significant results.
Results
Establishment and characterization of the
TMZ-resistant U87 cell line
The IC50 values of the U87 and TMZ-R U87
cells were 45 and >500 μM, respectively (Fig. 1A). The LN18 and T98G cells were more
chemoresistant to TMZ than the U87 parental cells. Morphologically,
TMZ-R U87 cells did not show a tendency for aggregation when they
reached subconfluency (Fig. 1B).
The activation (phosphorylation) of STAT3 and upregulation of MGMT
were identified in the TMZ-R U87 cell line compared with the U87
cell line using WB analysis (Fig.
1C). Additionally, the TMZ-R U87 cell line exhibited a greater
invasive activity compared with the parental U87 cell line
(Fig. 1D).
Characterization of U87-TMZR cell-derived
tumor xenografts in nude mice
The U87 parental and TMZ-R U87 cell lines were
transplanted into nude mice, and the sensitivity of the tumors to
TMZ was investigated. TMZ administration for 5 days showed a
significant inhibition on the parental U87 tumor growth and a
beneficial effect on the survival of tumor-bearing mice (Fig. 2A and B). In contrast, TMZ-R U87
cell-transplanted mice showed significant resistance to TMZ and a
shorter survival time in vivo (Fig. 2C and D).
Genetic profile and analysis of the
TMZ-resistant U87 cell line
Gene Chip microarray analysis demonstrated that the
expression of 10 genes was significantly altered (>5-fold) at
the 5th and 20th passages of the U87-TMZR cell line (upregulated:
CD24, YKL-40, GDF15, HLA-DQA1, MAGEC1, MGMT and MMP1;
downregulated: AMIGO2, NMU and RFC2) (Table I). qPCR showed that YKL-40 and
MAGEC1 were the top-2 upregulated genes (98- and 83-fold,
respectively) and 4 and 3 genes were >10-fold upregulated and
downregulated, respectively (Fig.
3). Meanwhile, as expected, MGMT expression was found to be
increased 6-fold, however its level was extremely low.
 | Table IFold-change of gene expression in the
TMZ-R U87 cell line compared to the U87 parental cell line. |
Table I
Fold-change of gene expression in the
TMZ-R U87 cell line compared to the U87 parental cell line.
| Gene symbol | ProbeSet ID | TMZ-R U87 (P5) | TMZ-R U87
(P20) |
|---|
| RFC2 | 1053_at | −5.1 | −5.5 |
| MMP-1 | 204475_at | 5.4 | 7.4 |
| NMU | 206023_at | −5.9 | −14.5 |
| MAGEC1 | 206609_at | 22.8 | 5.6 |
| YKL-40 | 209395_at | 12.6 | 6.6 |
| CD24 | 209771_x_at | 23.2 | 19.3 |
| HLA-DQA1/A | 2212671_s_at | 8.9 | 7.2 |
| GDF15 | 221577_x_at | 6.2 | 8.6 |
| AMIGO2 | 222108_at | −9.4 | −6.3 |
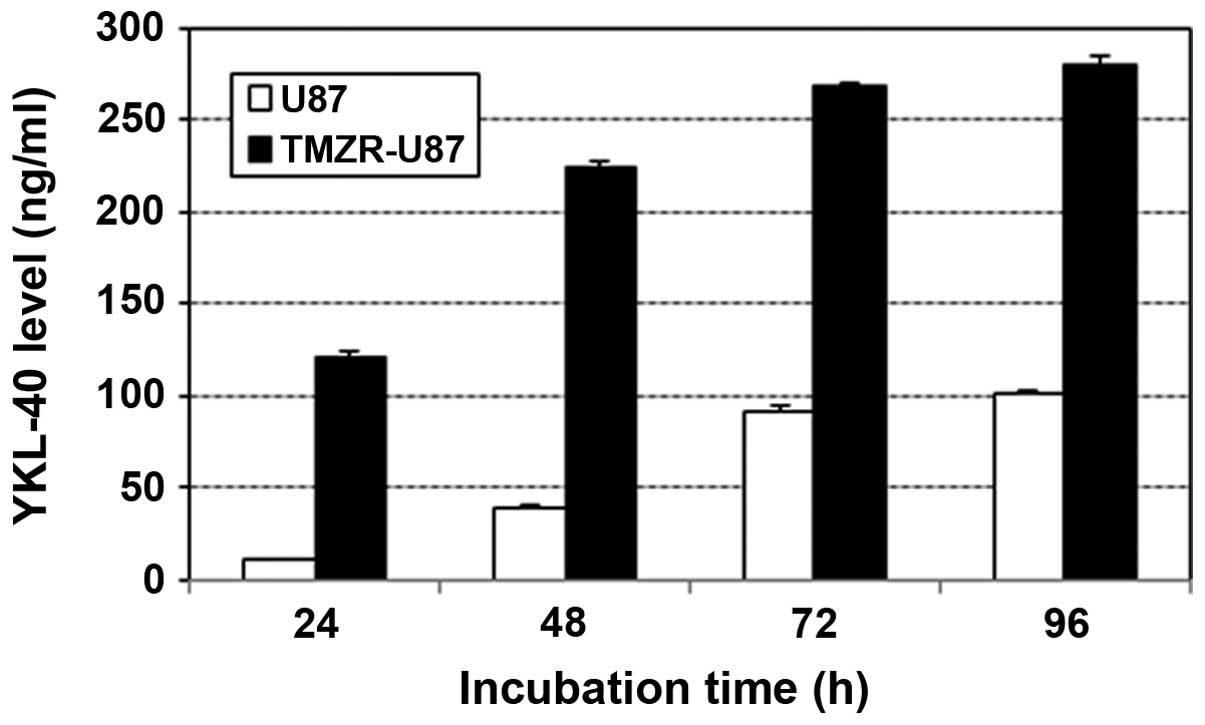
YKL-40 production in the TMZ-resistant
U87 cell line
The YKL-40 level in the supernatant of the TMZ-R U87
cell line was ~100 ng/ml at 24 h and reached >200 ng/ml at 48 h,
which were more than several fold upregulated compared with the
level of the parental U87 cell line (Fig. 4).
STAT3 target genes, glioma-associated
genes and EMT gene expression in the TMZ-resistant cell line
A >2-fold upregulation of many genes was
identified in the TMZ-R U87 cell line compared with the U87
parental cell line as follows: BCL2, Survivin, cMYC, p53 and HIF1A
as STAT3-target genes; ALDH1, GFAP, NANOG and SOX2 as stem cell
markers; FN-1, FOXC2, MMP2, SNAIL2, TCF4, TWIST1 and SMAD2 as
EMT-associated genes and STAT3 and C/EBPβ as mesenchymal genes
(Fig. 5).
Impact of YKL-40 inhibition on cell
proliferation, invasive activity and in vivo tumorigenesis in the
TMZ-resistant cell line
The YKL-40 protein levels in the shRNA-3 and
shRNA-4-transfected TMZ-R U87 cells were significantly reduced
(Fig. 6A). shRNA-mediated YKL-40
gene inhibition significantly suppressed the cell proliferation and
invasive activity of the TMZ-R U87 cells (Fig. 6B and C). Nude mice transplanted with
YKL-40 shRNA-4-transfected TMZ-R U87 cells showed significant
growth suppression compared with the mock gene-transfected TMZ-R
U87-transplanted mice after the 10th day of transplantation
(Fig. 6D).
Effect of YKL-40 or STAT3 gene inhibition
on the TMZ-resistance of TMZ-R U87 cells
TMZ-R U87 cells transfected with shRNA-4 exhibited
recovered sensitivity to TMZ at a dose of ~250 μM, which was
considered a partial effect compared with the parental U87
sensitivity to TMZ (Fig. 7A). In
contrast, TMZ-R U87 cells transfected with the mock gene showed no
sensitivity to TMZ, even at 1 mM. Additionally, STAT3 gene
inhibition by shRNA demonstrated a partially restorative effect on
TMZ-R U87 cells as well as YKL-40 gene inhibition (Fig. 7B).
Discussion
Glioblastoma multiforme (GBM) is one of the most
malignant tumors and has an extremely poor prognosis. Despite
recent therapeutic advances, the median survival of GBM patients is
less than one year, mainly since most cases relapse after
concomitant chemoradiation (1,2). Thus,
a novel therapeutic approach is urgently needed to control
recurrence and overcome resistance to treatment.
MGMT is well known to remove the methyl group from
the O6 position of guanine and contribute to TMZ
resistance, resulting in an important prognostic factor in the
clinical field (4). However, novel
therapeutic strategies that include
O6-benzylguanine to overcome the obtained TMZ
resistance have been attempted in clinical studies, but none have
been successful (5,6). Therefore, new approaches for
regulating MGMT expression and restoring the sensitivity to TMZ are
highly required.
In the present study, we demonstrated STAT3
activation, MGMT and YKL-40 upregulation in TMZ-R U87 cells, which
formed an in vivo transplanted tumor with obvious TMZ
resistance and YKL-40 high expression. The correlation of STAT3
with MGMT has been reported by Kohsaka et al, who
demonstrated that TMZ-R U87 cells exhibited active STAT3
phosphorylation and that STAT3 inhibition reduces MGMT expression
(23). These results suggest a
possible mechanism for MGMT regulation; however, it has not been
proven in in vivo experiments. On the other hand, Singh
et al reported that knockdown of STAT3 inhibited active
astrocyte migration through the reduction in YKL-40 production
(24). These results suggest that a
downstream pathway from STAT3 to MGMT through YKL-40 may exist and
contribute to the gain of TMZ resistance in GB tumors.
Additionally, the comprehensive qPCR study
demonstrated that many genes were upregulated in the TMZ-R U87 cell
line compared with the parental cell line. In particular, the
following gene groups appear to be involved in the malignant
features of TMZ-R U87 tumors: BCL-2, Survivin, c-Myc, Sox2 and
Nanog for tumorigenesis; CXCL10, FN1, MMP2, TWIST1 and SMAD2 for
invasive activity and ALDH1, STAT3 and C/EBPβ for TMZ resistance.
Thus, such machineries for causing TMZ resistance and an invasive
phenotype in clinical status will be challenging targets for
overcoming TMZ resistance and developing novel therapeutics against
TMZ-resistant GBM tumors.
YKL-40, also known as chitinase 3-like 1, human
cartilage glycoprotein 39, is a secreted glycoprotein that belongs
to the 18-glycosyl-hydrolase family of proteins. YKL-40 is produced
by many cell types including macrophages, neutrophils,
chondrocytes, smooth muscle and endothelial cells as well as
several types of cancer cells such as breast cancer, osteosarcoma,
ovarian cancer, lung cancer and GBM (25). YKL-40 is considered an angiogenic
factor for tumor vessel formation through VEGF production and
contributes to invasion and radio-resistance in in vivo
tumors (26,27).
YKL-40 is also a well-known biomarker for predicting
poor prognosis and a serum biomarker in GBM patients (28–33).
YKL-40 was reported as a prognostic marker in GBM patients for the
first time by Tanwar et al, who used DNA microarray analysis
(28). Similarly, several studies
(29–33) have demonstrated that YKL-40 is a
potential serum biomarker and prognostic marker of high-grade
glioma or other solid tumors such as ovarian and non-small cell
lung cancers. Notably, Bernardi et al reported that
postoperative YKL-40 level increases may be a negative prognostic
index by comparing serum YKL-40 levels before and after tumor
resection (34). However, the
involvement of YKL-40 in TMZ resistance has yet to be
clarified.
In the present study, we investigated the effect of
YKL-40 gene inhibition on TMZ resistance using shRNA gene
transfection into the TMZ-R U87 cell line. As a result, YKL-40 gene
inhibition significantly suppressed cell proliferation, invasive
activity and even tumorigenicity of TMZ-R U87 cells in vivo;
however, the restorative effect on TMZ resistance seemed to be
partial as well as STAT3 gene inhibition. Importantly, YKL-40 gene
inhibition did not induce the downregulation of MGMT expression
(data not shown), which may suggest a difference in the restoration
mechanism for TMZ sensitivity between YKL-40 and STAT3 gene
inhibition experiments.
YKL-40 is a good surrogate marker for STAT3
targeting since YKL-40 downregulation restores TMZ sensitivity and
suppresses TMZ-R tumor growth, which is a mechanism for overcoming
TMZ resistance. Therefore, YKL-40 is the next target by which
therapeutics against TMZ-resistant GB tumors can be developed. The
combination of a STAT3 inhibitor with an anti-YKL-40 antibody or
other YKL-40 inhibiting reagents could be a promising new approach
to overcome TMZ resistance in GB through suppression of
angiogenesis and invasion.
Acknowledgements
This study was supported by a grant from a regional
innovation strategy support program of the Ministry of Education,
Culture, Sports, Science and Technology, Japan.
Abbreviations:
|
GBM
|
glioblastoma multiforme
|
|
TMZ
|
temozolomide
|
|
STAT
|
signal transducer and activator of
transcription
|
|
SH
|
Src homology
|
|
DMSO
|
dimethyl sulfoxide
|
|
JAK
|
Janus kinase
|
|
T/C
|
tumor/control
|
|
siRNA
|
small interfering RNA
|
|
shRNA
|
small hairpin RNA
|
References
|
1
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mirimanoff RO, Gorlia T, Mason W, et al:
Radiotherapy and temozolomide for newly diagnosed glioblastoma:
recursive partitioning analysis of the EORTC 26981/22981-NCIC CE3
phase III randomized trial. J Clin Oncol. 24:2563–2569. 2006.
View Article : Google Scholar
|
|
3
|
Trivedi RN, Almeida KH, Fornsaglio JL,
Schamus S and Sobol RW: The role of base excision repair in the
sensitivity and resistance to temozolomide-mediated cell death.
Cancer Res. 65:6394–6400. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hegi ME, Diserens AC, Gorlia T, et al:
MGMT gene silencing and benefit from temozolomide in glioblastoma.
N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Quinn JA, Desjardins A, Weingart J, et al:
Phase I trial of temozolomide plus
O6-benzylguanine for patients with recurrent or
progressive malignant glioma. J Clin Oncol. 23:7178–7187.
2005.PubMed/NCBI
|
|
6
|
Quinn JA, Jiang SX, Reardon DA, et al:
Phase II trial of temozolomide plus
O6-benzylguanine in adults with recurrent,
temozolomide-resistant malignant glioma. J Clin Oncol.
27:1262–1267. 2009.PubMed/NCBI
|
|
7
|
Murat A, Migliavacca E, Gorlia T, et al:
Stem cell-related ‘self-renewal’ signature and high epidermal
growth factor receptor expression associated with resistance to
concomitant chemoradiotherapy in glioblastoma. J Clin Oncol.
26:3015–3024. 2008.
|
|
8
|
Hunter C, Smith R, Cahill DP, et al: A
hypermutation phenotype and somatic MSH6 mutations in recurrent
human malignant gliomas after alkylator chemotherapy. Cancer Res.
66:3987–3991. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun S, Lee D, Ho AS, et al: Inhibition of
prolyl 4-hydroxylase, beta polypeptide (P4HB) attenuates
temozolomide resistance in malignant glioma via the endoplasmic
reticulum stress response (ERSR) pathways. Neuro Oncol. 15:562–577.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mukherjee B, McEllin B, Camacho CV, et al:
EGFRvIII and DNA double-strand break repair: a molecular mechanism
for radioresistance in glioblastoma. Cancer Res. 69:4252–4259.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kitange GJ, Carlson BL, Schroeder MA, et
al: Expression of CD74 in high grade gliomas: a potential role in
temozolomide resistance. J Neurooncol. 100:177–186. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vlachostergios PJ, Hatzidaki E, Befani CD,
Liakos P and Papandreou CN: Bortezomib overcomes MGMT-related
resistance of glioblastoma cell lines to temozolomide in a
schedule-dependent manner. Invest New Drugs. 31:1169–1181. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kanzawa T, Germano IM, Kondo Y, Ito H, Kyo
S and Kondo S: Inhibition of telomerase activity in malignant
glioma cells correlates with their sensitivity to temozolomide. Br
J Cancer. 89:922–929. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng M, Bocangel D, Ramesh R, et al:
Interleukin-24 overcomes temozolomide resistance and enhances cell
death by down-regulation of O6-methylguanine-DNA
methyltransferase in human melanoma cells. Mol Cancer Ther.
7:3842–3851. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang G, Wei ZP, Pei DS, Xin Y, Liu YQ and
Zheng JN: A novel approach to overcome temozolomide resistance in
glioma and melanoma: Inactivation of MGMT by gene therapy. Biochem
Biophys Res Commun. 406:311–314. 2011. View Article : Google Scholar
|
|
16
|
Tso CL, Shintaku P, Chen J, et al: Primary
glioblastomas express mesenchymal stem-like properties. Mol Cancer
Res. 4:607–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Phillips HS, Kharbanda S, Chen R, et al:
Molecular subclasses of high-grade glioma predict prognosis,
delineate a pattern of disease progression, and resemble stages in
neurogenesis. Cancer Cell. 9:157–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Carro MS, Lim WK, Alvarez MJ, et al: The
transcriptional network for mesenchymal transformation of brain
tumors. Nature. 463:318–325. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Verhaak RG, Hoadley KA, Purdom E, et al:
Integrated genomic analysis identifies clinically relevant subtypes
of glioblastoma characterized by abnormalities in PDGFRA, IDH1,
EGFR, and NF1. Cancer Cell. 17:98–110. 2010. View Article : Google Scholar
|
|
20
|
Ashizawa T, Miyata H, Iizuka A, et al:
Effect of the STAT3 inhibitor STX-0119 on the proliferation of
cancer stem-like cells derived from recurrent glioblastoma. Int J
Oncol. 43:219–227. 2013.PubMed/NCBI
|
|
21
|
Ashizawa T, Miyata H, Ishii H, et al:
Antitumor activity of a novel small molecule STAT3 inhibitor
against a human lymphoma cell line with high STAT3 activation. Int
J Oncol. 38:1245–1252. 2011.PubMed/NCBI
|
|
22
|
Workman P, Aboagye EO, Balkwill F, Balmain
A, Bruder G, Chaplin DJ, Double A, Everitt J, Farningham DAH,
Glennie MJ, Kelland LR, Robinson V, Stratford IJ, Tozer GM, Watson
S, Wedge SR and Eccles SA: An ad hoc committee of the National
Cancer Research Institute: Guidelines for the welfare and use of
animals in cancer research. Br J Cancer. 102:1555–1577. 2010.
View Article : Google Scholar
|
|
23
|
Kohsaka S, Wang L, Yachi K, et al: STAT3
inhibition overcomes temozolomide resistance in glioblastoma by
downregulating MGMT expression. Mol Cancer Ther. 11:1289–1299.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Singh SK, Bhardwaj R, Wilczynska KM, Dumur
CI and Kordula T: A complex of nuclear factor I-X3 and STAT3
regulates astrocyte and glioma migration through the secreted
glycoprotein YKL-40. J Biol Chem. 286:39893–39903. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kzhyshkowska J, Gratchev A and Goerdt S:
Human chitinases and chitinase-like ptoteins as indicators for
inflammation and cancer. Biomark Insights. 3:128–146.
2007.PubMed/NCBI
|
|
26
|
Shao R, Hamel K, Petersen L, et al:
YKL-40, a secreted glycoprotein, promotes tumor angiogenesis.
Oncogene. 28:4456–4468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Francescone RA, Scully S, Faibish M, et
al: Role of YKL-40 in the angiogenesis, radioresistance, and
progression of glioblastoma. J Biol Chem. 286:15332–15343. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tanwar MK, Gilbert MR and Holland EC: Gene
expression microarray analysis reveals YKL-40 to be a potential
serum marker for malignant character in human glioma. Cancer Res.
62:4364–4368. 2002.
|
|
29
|
Høgdall EV, Johansen JS, Kjaer SK, et al:
High plasma YKL-40 level in patients with ovarian cancer stage III
is related to shorter survival. Oncol Rep. 10:1535–1538.
2003.PubMed/NCBI
|
|
30
|
Hormigo A, Gu B, Karimi S, et al: YKL-40
and matrix metalloproteinase-9 as potential serum biomarkers for
patients with high-grade gliomas. Clin Cancer Res. 12:5698–5704.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thöm I, Andritzky B, Schuch G, et al:
Elevated pretreatment serum concentration of YKL-40 - An
independent prognostic biomarker for poor survival in patients with
metastatic nonsmall cell lung cancer. Cancer. 116:4114–4121.
2010.
|
|
32
|
Zhang W, Kawanishi M, Miyake K, et al:
Association between YKL-40 and adult primary astrocytoma. Cancer.
116:2688–2697. 2010.PubMed/NCBI
|
|
33
|
Iwamoto FM, Hottinger AF, Karimi S, et al:
Serum YKL-40 is a marker of prognosis and disease status in
high-grade gliomas. Neuro Oncol. 13:1244–1251. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bernardi D, Padoan A, Ballin A, et al:
Serum YKL-40 following resection for cerebral glioblastoma. J
Neurooncol. 107:299–305. 2012. View Article : Google Scholar : PubMed/NCBI
|