Introduction
The human defensins are small cationic antimicrobial
peptides that are classified into two classes, α-and β-defensins.
The α-defensins are expressed in neutrophils and in Paneth cells in
the intestinal crypt. Human β-defensins (hBDs) are mainly produced
by the epithelial cells of numerous organs including the skin,
lung, kidney, pancreas, uterus, eye and nasal and oral mucosa.
hBD-1, -2 and -3 among the many types of hBDs have been well
researched. hBD-1 is constitutively expressed by various tissues
and may be modulated by inflammation. hBD-2 and hBD-3 are expressed
by cells upon stimulation with proinflammatory cytokines such as
interleukin (IL)-1β, tumor necrosis factor (TNF)-α and interferon
(IFN)-γ and by microorganisms (1,2).
Evidence has recently suggested the involvement of hBDs in
carcinogenesis as well as their antimicrobial activity (3,4). The
expression levels of hBD-1 and hBD-2 mRNA were found to be lower in
oral carcinoma than in healthy oral epithelium (5–7).
Overexpression of β-defensin-1 in renal carcinoma cells induced
apoptotic cell death, implying that hBD-1 may function as a
tumor-suppressor gene (8).
Decreased expression of hBD-1 in OCCs may be due to their single
nucleotide polymorphisms (SNPs) (7), and may play a role in the development
of oral carcinoma (6). It is,
however, still unclear how hBD-2 expression is downregulated in
oral carcinoma. Gene polymorophisms and mutations, loss of
heterozygosity, and epigenetic modifications are involved in the
aberrant transcriptional levels in malignant tumors (9,10).
Neither directly connected SNPs, common gene mutations nor
epigenetic modifications of hBD-2 have been shown to date. DNA
methylation and histone modifications are two major mechanisms of
epigenetic modifications in human cells (11). DNA methylation is characterized by
the addition of a methyl group to cytosines within CpG regions. DNA
hypermethylation leading to a decrease in gene transcriptional
levels frequently occurs in tumor-suppressor genes in malignant
tumors. The decreased expression of tumor-suppressor genes by DNA
hypermethylation promotes malignant transformation (9–11).
Therefore, we hypothesized that DNA hypermethylation
is involved in the decreased expression of hBD-2 in OCCs, and hBD-2
may play a role in the development of OCCs. The present study
investigated the DNA hypermethylation of hBD-2 in OCCs and the
effect of demethylation and increased expression of hBD-2 on cell
proliferation and invasion.
Materials and methods
Cell cultures
Normal human oral keratinocytes (NOKs) were isolated
from healthy gingival tissue overlying the impacted third molar of
an adult human. Briefly, explants of the healthy gingival tissue
obtained from the third molar surgical extraction were cultured in
Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich, St. Louis,
MO, USA) containing 10% fetal bovine serum (FBS; Gibco, Invitrogen
Corp., Carlsbad, CA, USA) and antibiotics (100 U/ml penicillin; 200
μg/ml streptomycin; 5 μg/ml amphotericin B; Sigma-Aldrich) and 30
mg/ml Fungizone (Bristol-Myers Squibb, Tokyo, Japan) at 37°C in a
humidified atmosphere of 95% air and 5% CO2. Outgrowth
developed after 2 or 3 weeks of incubation. The two cell types were
separated into epithelial populations that were more or less
resistant to detachment with 10% dispase (Godo Shusei Co., Ltd.,
Tokyo, Japan). The cells that were less resistant were detached,
removed from the rest of the cell population and discarded. The
attached cells were cultured. The separation procedure was repeated
two or three times so as to remove any fibroblasts. NOKs were grown
in Keratinocyte Basal Medium (KBM; Lonza Walkersville, Inc.,
Walkersville, MD, USA) supplemented with 7.5 mg/ml bovine pituitary
extract (Lonza), 0.1 μg/ml hEGF (Lonza), 5 mg/ml insulin (Lonza),
0.5 ml aqueous solution of gentamicin sulfate (Lonza) and
amphotericin-B.
Five different types of oral squamous cell carcinoma
cell lines (OSC-19, BSC-OF, SAS, HSC-2 and HSC-4) and a human
salivary adenocarcinoma cell line (HSY) were incubated with DMEM
supplemented with 10% FBS, 100 U/ml penicillin and 200 μg/ml
streptomycin.
To examine the role of hBD-2 methylation in OCCs,
the cells were treated with 50 μM of the DNA demethylating agent,
5-aza-2′-deoxycytidine (5-aza-dC; Sigma-Aldrich) or the same volume
of DMSO as control for 24 and 48 h.
Quantitative real-time RT-PCR
The expression levels of hBD-2 mRNA in the cultured
cells were analyzed by quantitative real-time RT-PCR. Total RNA was
extracted from cells by the acid guanidine
thiocyanate/phenol-chloroform method using TRIzol (Invitrogen). The
RNA was reverse transcribed (SuperScript Reverse Transcriptase;
Invitrogen), according to the manufacturer’s instructions using
oligo(dT)12–18 primers (Invitrogen). Specific primer and
probe sets for hBD-2 and GAPDH were purchased from Applied
Biosystems (Foster City, CA, USA) (hBD-2; Hs00823638_m1, GAPDH;
Mm99999915_g1). The reaction mixture was prepared using TaqMan
Universal PCR Master Mix (Applied Biosystems) with primer, probe
sets and RT products. Real-time PCR was performed using a GeneAmp
5700 Sequence Detection System instrument and software (Applied
Biosystems). The expression level of hBD2 mRNA was standardized
against GAPDH mRNA. The relative expression of hBD-2 mRNA was
calculated using the 2−ΔΔCt method as described by
Saitoh et al (12). Data are
expressed as the ratio of hBD-2 mRNA to GAPDH mRNA.
Methylation-specific polymerase chain
reaction method
In order to analyze CpG island hypermethylation of
hBD-2, the methylation profiles were assessed using a
methylation-specific PCR (MSP) method, similar to that reported by
Herman et al (13). Briefly,
DNA was extracted from the cultured cells with DNeasy Blood &
Tissue kit (Qiagen, Stanford, CA, USA) according to the
manufacturer’s protocol. The DNA was purified using a
phenol/ethanol method. MSP distinguishes unmethylated from
methylated alleles in a given gene based on sequence changes
produced after bisulfite treatment of DNA, which converts
unmethylated (but not methylated) cytosines to uracil, and
subsequent PCR using primers designed for either methylated or
unmethylated DNA. The primer sequences are listed in Table I. PCR was performed using an
amplification kit (AmpliTag Gold; Applied Biosystems) and a thermal
cycler (Takara PCR Thermal Cycler MP; Takara, Osaka, Japan). Each
PCR product was loaded directly onto non-denaturing 2% agar gels.
As a positive control for methylation, CpGenome™ Universal
Methylation DNA (Millipore, Billerica, MA, USA) was used.
 | Table IhBD2 primer design for MSP. |
Table I
hBD2 primer design for MSP.
| Primers 241 bp | Primer sequence
(5′-3′) |
|---|
| Methylation primers
(M) | F:
TTGGTTTGTTAGGAATTAGGGTTT |
| R:
CCATCCCGAACACTCAAAA |
| Unmethylation primers
(U) | F:
TTGGTTTGTTAGGAATTAGGGTTT |
| R:
CCATCCCAAACACTCAAAAA |
Increased expression of hBD-2
hBD-2 containing the entire coding region was
amplified by PCR using NOK cDNA as a template. The primer sets used
were 5′-CGCGGATCCATGA GGGTCTTGTAT-3′ (forward) and 5′-CCGCTCGAGTCAT
GGCTTTTTGCA-3′ (reverse) for cDNA amplification. The amplified
products were inserted into BamH1 and XhoI cloning
sites of the pcDNA 6/His C (Invitrogen). The inserted hBD-2
sequence was confirmed with a standard DNA sequencing method. SAS
cells were transfected with the hBD-2-inserted plasmid using the
lipofection method (Effecten™; Qiagen, Hilden, Germany).
Transfected clones were selected in 5 μg/ml Blasticidin S
(Invitrogen). As a control, the pcDNA 6/His C mock vector was
transfected into the same cell lines.
Cell proliferation assay
Control and hBD-2-transfected SAS cells were seeded
at a concentration of 2×105 cells/ml on 96-well plates
in DMEM containing 10% FBS and were cultured for 24 h. After 24 and
48 h of incubation of the cells, XTT assays were performed. XTT
solution (1 mg/ml XTT in 80 ml; Sigma) and 0.025 mM phenazine
methosulphate were added to the cells in a 96-well microplate.
After a 3-h incubation, the optical density (OD) at 490 nm was
measured.
Cell invasion assay
Cell migration was assessed in 6-well Transwell
units with an 8-μm pore polycarbonate filter membrane coated with
Matrigel™ (BD Biosciences, San Jose, CA, USA). Control and
hBD-2-transfected SAS cells in serum-free DMEM at a density of
2×105 cells/ml were loaded on the membrane in the upper
chamber. The upper chambers were gently inserted into DMEM
supplemented with 10% FBS in the lower chamber. After 7 days,
non-migrating cells on the upper side of the membrane were removed
by a sterile cotton swab, and the remaining invaded cells on the
lower side of the membrane were fixed and stained with hematoxylin.
The number of cells in three fields per well were counted under a
microscope.
Statistical analysis
Data from the real-time RT-PCR, cell proliferation
assay and cell mobility assay were analyzed using the Student’s
t-test (2-tailed). A P-value of <0.05 was considered to indicate
a statistically significant result.
Results
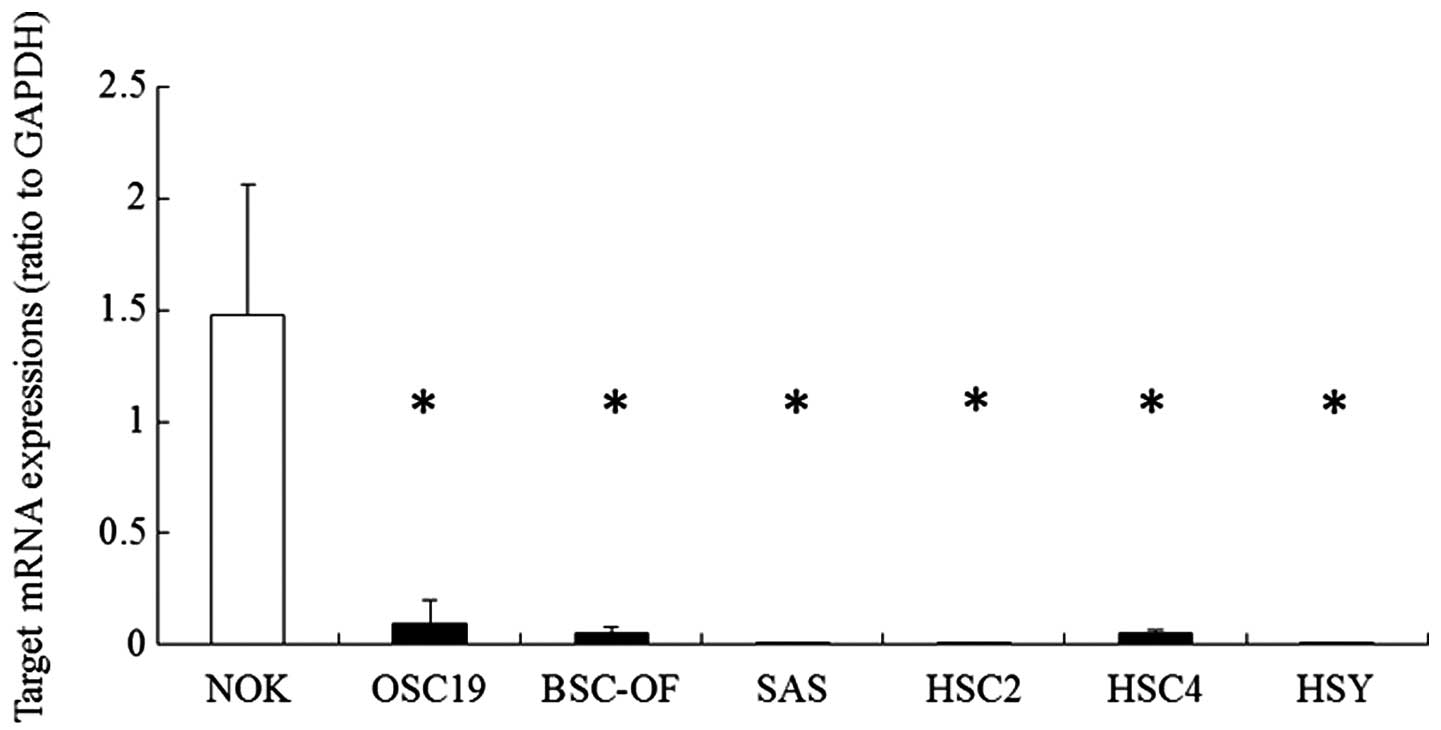
First, we observed the expression of hBD-2 mRNA in
the OCCs by quantitative RT-PCR. The expression levels in OCCs were
compared with that in the NOKs. The expression levels of hBD-2 mRNA
in the OCCs (OSC-19, BSC-OF, SAS, HSC-2, HSC-4 and HSY) were
significantly lower than that in the NOKs (Fig. 1). In order to examine whether DNA
hypermethylation is involved in the transcriptional levels, cells
were treated with DNA methyltransferase inhibitor, 5-aza-dC, at the
concentration of 50 μM. The inhibitor significantly induced
upregulated expression of hBD-2 in the OCCs (Fig. 2). The results indicate that DNA
hypermethylation may be involved in the downregulated expression of
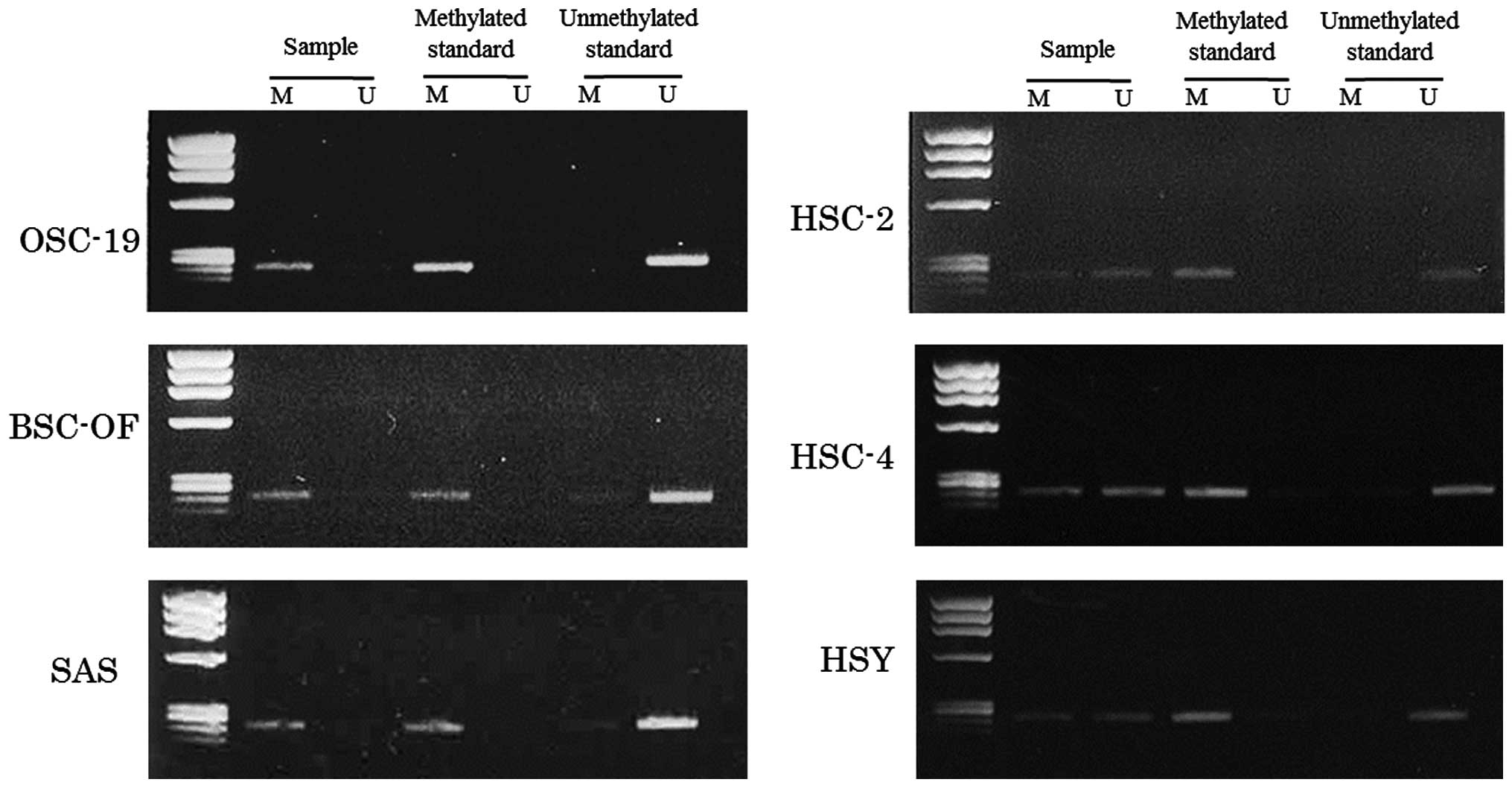
hBD-2 mRNA in the OCCs. The DNA hypermethylation in the OCCs was
examined using an MSP method. The primers for the MSP method were
designed in the promoter region lower than −1,500 kbp. The data for
the MSPs are illustrated in Fig. 3.
Bisulfite treatment converts unmethylated cytosines to uracil,
which shows a visible PCR product when amplified by unmethylated
primers (lane U). No conversion of the methylated cytosines was
shown as a visible PCR product when amplified by methylated primers
(lane M). The product visualized only in lane M, both in U and M,
and only in U indicates complete, partial and no hypermethylation,
respectively. Complete hypermethylation was observed in OSC-19,
BSC-OF and SAS cells, and partial hypermethylation was observed in
HSC-2, HSC-4 and HSY cells (Fig.
3). These results confirmed that DNA hypermethylation was, at
least in part, involved in the decreased expression of hBD-2 in the
OCCs.
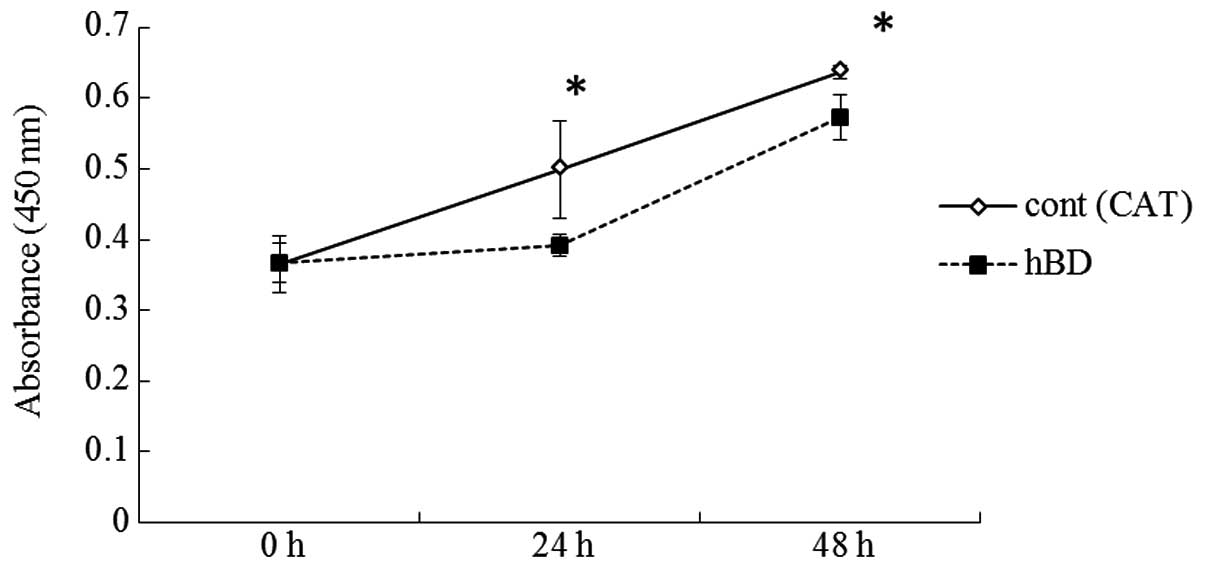
In order to ascertain whether the decreased
expression of hBD-2 is involved in cell proliferation and invasion
implying malignant potential, we examined whether 5-aza-dC that
induced upregulated expression of hBD-2 in cells could inhibit the
cell proliferation and invasion of OCCs. The growth rates of SAS,
HSC-2, HSC-4 and HSY cells treated with 5-aza-dC at the
concentration of 50 μM were significantly lower than that of the
controls. No significant differences were observed between the
OSC-19 or BSC-OF cells treated with 5-aza-dC and the controls
(Fig. 4). The cell invasion assays
showed that the number of OCCs treated with 5-aza-dC on the filters
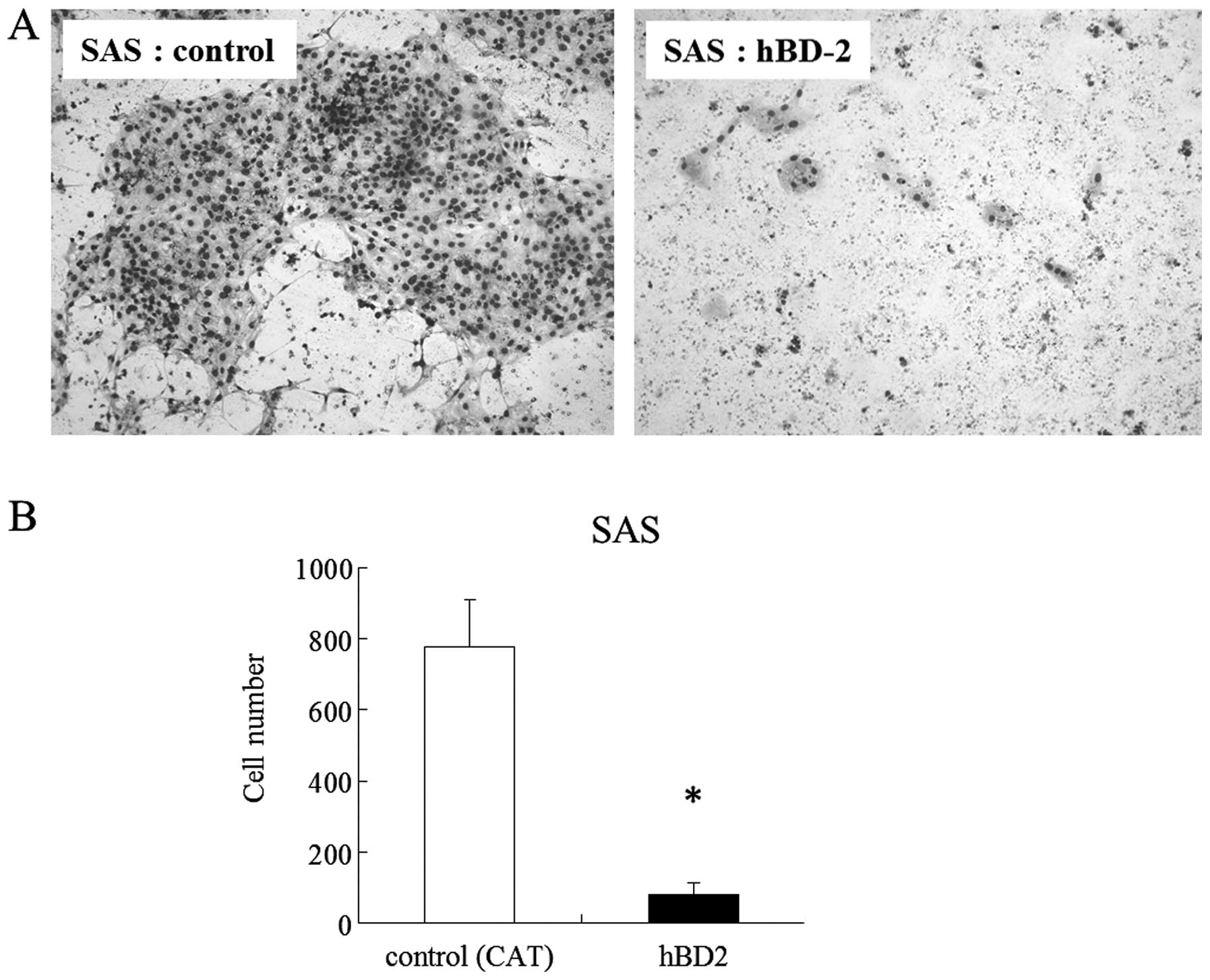
were significantly less than that of the controls (Fig. 5). We examined whether increased
expression of hBD-2 generated by gene transfection inhibited the
cell proliferation and invasion. Since expression level of hBD-2 in
SAS cells was lowest among the OCCs, increased expression of hBD2
in SAS was generated by the hBD-2 gene. The number of cells
exhibiting increase expression of hBD-2 were significantly lower at
24 and 48 h than the control (Fig.
6). The number of cells exhibiting increased expression of
hBD-2 that migrated to the filters in the invasion assay were
significantly lower on day 7 than the control (Fig. 7).
Discussion
The present study first demonstrated that DNA
hypermethylation is involved in decreased expression of hBD-2 in
OCCs, and that increased expression of hBD-2 inhibited OCC
proliferation and invasion. Thus, hBD-2 is likely to be a putative
tumor-suppressor gene.
The expression levels of hBD-2 mRNA were found to be
lower in OCCs than that in healthy oral epithelium (5,7,14). The
present study showed that the expression level of hBD-2 in 6 types
of OCCs was lower than that in NOKs which supported the previous
data. The aberrant expression of certain types of genes are
frequently observed in cancers. The aberrant expression of
tumor-suppressor genes such as p14, p15, p16 and p53 is often
involved in carcinogenesis (9,15).
Several point mutations frequently occur in p53. The half-life of
mutant p53 is longer than that of the wild-type, resulting in the
accumulation of mutant p53. The accumulation leads to
overexpression of p53 (16). The
p14, p15 and p16 genes are often downregulated induced by DNA
hypermethylation in malignant cells. DNA hypermethylation in p14,
p15 and p16 was frequently found in OCCs (9,10).
Therefore, we examined whether the downregulation of the expression
of hBD-2 involves DNA hypermethylation. Treatment with 5-aza-dC, a
DNA methyltransferase inhibitor, significantly induced the
upregulation of the expression of hBD-2 in the OCCs. The MSP method
confirmed the DNA hypermethylation of hBD2 in the OCCs. These
results suggest that DNA hypermethylation is involved in the
decreased expression of hBD-2 in OCCs.
As mentioned above, tumor-suppressor genes such as
p14, p15 and p16 are often downregulated in OCCs, induced by DNA
hypermethylation. The downregulation is possibly linked to OCC
development (9,10). hBD-2 may function as a
tumor-suppressor since downregulated expression of hBD-2 is caused
by DNA hypermethylation in OCC. In order to clarify this
speculation, we examined whether increased expression of hBD-2 in
the OCCs affects their cell proliferation and invasion capacities.
Treatment with 5-aza-dC inhibited the cell proliferation and
invasion of OCCs as well as induced upregulated expression of
hBD-2. DNA methyltransferase inhibitors including azacytidene,
decitabine and zabularine have been widely applied for experimental
cancer therapy (17). Our data
confirmed that 5-aza-dC, a type of azacytidene, may be a
therapeutic agent for OC. It was unclear whether the inhibition of
cell proliferation and invasion were due to the effect of 5-aza-dC
or the increased expression of hBD-2. We observed the effect of
increased expression of hBD-2 on OCC proliferation and invasion.
Since the expression level of hBD-2 in SAS cells was lowest among
the OCCs, we used SAS cells transfected by the hBD-2 gene to
increase the expression of hBD-2. Increased expression of hBD-2 in
the SAS cells inhibited their proliferation and invasion. The
inhibitory effect of 5-aza-dC on OCC proliferation and invasion may
be partially via increased expression of hBD-2. The effects of the
hBD-2 peptide on carcinoma cell lines have been previously reported
(18–20). hBD-2 was found to have little effect
on the proliferation of carcinoma cells (18). The hBD-2 peptide was reported to
promote and inhibit the proliferation of carcinoma cells at low and
high concentrations, respectively (19). hBD-2 promoted the proliferation of
carcinoma cells, implying its function as a proto-oncogene
(20). The effects of hBD-2 on
carcinoma cells may be dependent on the concentration of the hBD-2
peptide and the type of cells. Unlike these previous reports that
showed an effect of the exogenous hBD-2 peptide on the cells, we
observed the effect of endogenous hBD-2 expression on carcinoma
cells. The increased endogenous expression of hBD-2 clearly
inhibited their own cell proliferation and invasion. The mechanism
of the inhibitory effect of endogenous hBD-2 on carcinoma cells is
unknown. The inhibitory effect of exogenous hBD-2 peptide on cell
proliferation might be via G1/S arrest and pRB gene expression
(19). The increased expression of
hBD-1 in renal carcinoma cells generated by hBD-1 gene transfection
inhibited proliferation of their own cells and resulted in
caspase-3-mediated apoptosis (8).
The overexpression of hBD-1 in a keratinocyte cell line, HaCaT,
induced by gene transfection promoted differentiation of the cells.
On the other hand, the overexpression of hBD-2 in the HaCaT cells
did not affect their differentiation (21). Although no increased expression of
hBD-2 was observed in the HaCaT cells under the conditions required
for differentiation (22), the
expression of hBD-2 was upregulated in other types of keratinocytes
by stimulation with keratinocyte differentiation (23,24).
An inhibitory effect of increased expression of hBD-2 on the
proliferation and invasion of OCCs was noted which may be due to
the response to stimulation of OCC differentiation. Further
investigations are needed to clarify the inhibitory mechanism of
cell proliferation and invasion by endogenous hBD-2 expression. The
inhibitory effect caused by increased endogenous expression of
hBD-1 on the carcinoma cells suggests that hBD-1 is a potential
tumor-suppressor gene. Further investigations will be needed to
verify this speculation.
In conclusion, we found involvement of DNA
hypermethylation in the decreased expression of hBD-2 in oral
carcinoma cell lines. Increased expression of hBD-2 in the cells
inhibited their proliferation and invasion. Therefore, hBD-2
functions as a tumor suppressor. Increased expression of hBD-2
induced by demethylation or by gene transfection may be a useful
therapeutic method for oral carcinoma.
References
|
1
|
Abiko Y and Saitoh M: Salivary defensins
and their importance in oral health and disease. Curr Pharm Des.
13:3065–3072. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Diamond G and Ryan L: Beta-defensins: what
are they really doing in the oral cavity? Oral Dis. 17:628–635.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Droin N, Hendra JB, Ducoroy P and Solary
E: Human defensins as cancer biomarkers and antitumour molecules. J
Proteomics. 72:918–927. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Semple F and Dorin JR: β-Defensins:
multifunctional modulators of infection, inflammation and more? J
Innate Immun. 4:337–348. 2012.
|
|
5
|
Abiko Y, Mitamura J, Nishimura M,
Muramatsu T, Inoue T, Shimono M and Kaku T: Pattern of expression
of beta-defensins in oral squamous cell carcinoma. Cancer Lett.
23:37–43. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wenghoefer M, Pantelis A, Dommisch H,
Reich R, Martini M, Allam JP, et al: Decreased gene expression of
human β-defensin-1 in the development of squamous cell carcinoma of
the oral cavity. Int J Oral Maxillofac Surg. 37:660–663. 2008.
|
|
7
|
Joly S, Compton LM, Pujol C, Kurago ZB and
Guthmiller JM: Loss of human β-defensin 1, 2, and 3 expression in
oral squamous cell carcinoma. Oral Microbiol Immunol. 24:353–360.
2009.
|
|
8
|
Sun CQ, Arnold R, Fernandez-Golarz C,
Parrish AB, Almekinder T, He J, et al: Human β-defensin-1, a
potential chromosome 8p tumor suppressor: control of transcription
and induction of apoptosis in renal cell carcinoma. Cancer Res.
66:8542–8549. 2006.
|
|
9
|
González-Ramírez I, García-Cuellar C,
Sánchez-Pérez Y and Granados-García M: DNA methylation in oral
squamous cell carcinoma: molecular mechanisms and clinical
implications. Oral Dis. 17:771–778. 2011.PubMed/NCBI
|
|
10
|
Radhakrishnan R, Kabekkodu S and
Satyamoorthy K: DNA hypermethylation as an epigenetic mark for oral
cancer diagnosis. J Oral Pathol Med. 40:665–676. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen QW, Zhu XY, Li YY and Meng ZQ:
Epigenetic regulation and cancer (Review). Oncol Rep. 31:523–532.
2014.PubMed/NCBI
|
|
12
|
Saitoh M, Kurashige Y, Yamazaki M,
Nishimura M, Nakamura S, Noro D, et al: Increased expression of
β-defensin-2 and -3 during the development of autoimmune
sialoadenitis in MRL/lpr mice. Med Mol Morphol. 40:157–162.
2007.
|
|
13
|
Herman JG, Graff JR, Myöhänen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: a novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Winter J, Pantelis A, Reich R, et al:
Human beta-defensin-1, -2, and -3 exhibit opposite effects on oral
squamous cell carcinoma cell proliferation. Cancer Invest.
29:196–201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Macaluso M, Montanari M, Cinti C and
Giordano A: Modulation of cell cycle components by epigenetic and
genetic events. Semin Oncol. 32:452–457. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim E and Deppert W: Transcriptional
activities of mutant p53: when mutations are more than a loss. J
Cell Biochem. 93:878–886. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gnyszka A, Jastrzebski Z and Flis F: DNA
methyltransferase inhibitors and their emerging role in epigenetic
therapy of cancer. Anticancer Res. 33:2989–2996. 2013.PubMed/NCBI
|
|
18
|
Nishimura M, Abiko Y, Kurashige Y,
Takeshima M, Yamazaki M, Kusano K, Saitoh M, Nakashima K, Inoue T
and Kaku T: Effect of defensin peptides on eukaryotic cells:
primary epithelial cells, fibroblasts and squamous cell carcinoma
cell lines. J Dermatol Sci. 36:87–95. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhuravel E, Shestakova T, Efanova O,
Yusefovich Y, Lytvin D, Soldatkina M and Pogrebnoy P: Human
beta-defensin-2 controls cell cycle in malignant epithelial cells:
in vitro study. Exp Oncol. 33:114–120. 2011.PubMed/NCBI
|
|
20
|
Winter J, Pantelis A, Reich R, Martini M,
Kraus D, Jepsen S, Allam JP, Novak N and Wenghoefer M: Human
α-defensin (DEFA) gene expression helps to characterise benign and
malignant salivary gland tumours. BMC Cancer. 12:4652012.
|
|
21
|
Frye M, Bargon J and Gropp R: Expression
of human beta-defensin-1 promotes differentiation of keratinocytes.
J Mol Med. 79:275–282. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Abiko Y, Nishimura M, Kusano K, Yamazaki
M, Arakawa T, Takuma T and Kaku T: Upregulated expression of human
β defensin-1 and -3 mRNA during differentiation of keratinocyte
immortalized cell lines, HaCaT and PHK16-0b. J Dermatol Sci.
31:225–228. 2003.
|
|
23
|
Liu AY, Destoumieux D, Wong AV, Park CH,
Valore EV, Liu L and Ganz T: Human β-defensin-2 production in
keratinocytes is regulated by interleukin-1, bacteria, and the
state of differentiation. J Invest Dermatol. 118:275–281. 2002.
|
|
24
|
Harder J, Meyer-Hoffert U, Wehkamp K,
Schwichtenberg L and Schröder JM: Differential gene induction of
human β-defensins (hBD-1, -2, -3, and -4) in keratinocytes is
inhibited by retinoic acid. J Invest Dermatol. 123:522–529.
2004.
|