Introduction
The annual incidence of gliomas which account for
more than 50% of all brain tumors in adults is 6/100,000 (1). Based on the histopathologic subgroups
characterized by different levels of aggressiveness and malignancy,
astrocytomas account for 60–70% of gliomas. Extensive surgical
resection, which correlates with patient survival independently
(2,3), followed by radiotherapy is one of the
standard treatments for glioma. Yet, the prognosis remains dismal
due to the ability of gliomas to infiltrate diffusely into the
normal brain parenchyma, a direct consequence of the transformation
into genetic higher-grade gliomas and recurrence (4–6). As
known, radioresistance is a common phenomenon in gliomas and, to
date there are no valid biomarkers for evaluating
radiosensitivity.
In fact, radiotherapy which relies on the generation
of oxygen super-radicals, often fails to kill tumor cells, due to
inadequate oxygen stress within the tumor cell mass (7,8).
However, in tumor microenvironments where oxygen is scarce and
glucose consumption is high, as within the expanding tumor mass, a
metabolic pathway shift occurs in order to maintain local
homeostasis. This phenomenon is known as the Warburg effect.
Transcription of numerous enzymes such as phosphofructokinase-1
(PFK-1), lactate dehydrogenase (LDH) and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) is activated in the glycolytic pathway. Among
these enzymes, PGK1, an ATP-generating glycolytic enzyme, plays a
vital role. It catalyzes the transfer of a high-energy phosphoryl
group from the acyl phosphate of 1,3-diphosphoglycerate to ADP to
produce ATP. PGK1 has also been proven to have an increased
expression level in many malignant tumors (9–16). The
possible role of PGK1 in radioresistance has been previously
studied. Data from our laboratory showed that expression of PGK1 is
significantly upregulated in radioresistant astrocytomas and
radioresistant glioma U251 cells (17,18).
As an initial step toward identification of a clinically applicable
method for regulating PGK1 for combination with radiotherapy, we
investigated the effects of shRNA-PGK1 on the in vitro
radiosensitivity of human glioma U251 cells (18). Downregulation of PGK1 by shRNA
combined with radiotherapy significantly decreased cell viability
and reduced the ability of migration and invasion in U251 cells.
This indicates that PGK1 may be closely associated with glioma
radioresponse and may be a potential target with which to sensitize
conventional radiotherapy. However, the exact effect of PGK1 on the
radiosensitivity in vivo using animal models has not yet
been clarified. To further evaluate the potential radioresistance
of PGK1, we extended these in vitro results to an in
vivo xenograft model. Mice bearing U251 xenografts were exposed
to shRNA-PGK1 and the level of PGK1 was determined. Radiation was
then delivered in a single exposure, and shRNA-PGK1 treatment
resulted in a significant enhancement in tumor response. These
findings suggest a possible approach for the rational design of a
clinical protocol combining downregulation of PGK1 and
radiotherapy.
Materials and methods
Cell lines and culture
Human U251 cells were obtained from KeyGen Biotech
(Nanjing, China), and radioresistant U251 cells (RR-U251 cells)
were established according to a previously described method
(18). The cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10%
fetal calf serum (both from Gibco-Invitrogen, Carlsbad, CA, USA),
100 U/ml of penicillin and 100 μg/ml of streptomycin in a 5%
CO2 humidified incubator at 37°C.
Plasmid molecules and stable transfection
in vitro
The shRNA-PGK1 plasmid and the high expression
plasmid pcDNA3.1-PGK1 were synthesized commercially by GenePharma
Co. Ltd. (Shanghai, China). shRNA1 (5′-GCAAGGATGTTCTGTTCTTGA-3′),
shRNA2 (5′-GCTCAACAACATGGAGATTGG-3′), shRNA3
(5′-GGATGTCTATGTCAATGATGC-3′) and shRNA4
(5′-GAAGATTACCTTGCCTGTTGA-3′) were designed to target the coding
region of the human PGK1 sequence (gene ID: 5230). A negative
control shRNA-NC (5′-GTTCTCCGAACGTGTCACGT-3′) was also obtained
from Shanghai GenePharma. U251 cells were trypsinized and harvested
from a monolayer to a cell suspension in DMEM containing 10% fetal
bovine serum without antibiotics and allowed to seed overnight.
When the cell confluency was 70–80%, complexes of shRNAs or
pcDNA3.1-PGK1 duplexes and Lipofectamine 2000 (Invitrogen) were
prepared as follows: 4 μg shRNAs or pcDNA3.1-PGK1 and 8 μl of
Lipofectamine 2000 were diluted in 240 μl Opti-MEM I medium (Gibco)
separately, and after 5 min standing at room temperature, gentle
mixing was conducted. After a 20-min incubation of this mixture,
the complexes were added to each well (final concentration for DNA
plasmids was 2 μg/ml). The medium for transfection was replaced by
complete medium at 6 h after transfection and observed under a
fluorescence microscope (Axiovert 40 CFL; Carl Zeiss, Oberkochen,
Germany) to observe the transfection efficiency at 24 h after
transfection. The expression of PGK1 was analyzed, respectively, at
24 and 48 h after transfection. A control and a negative control
were also placed in the 6-well plates. In addition, complete medium
with G418 (400 μg/ml) (Keygen, Nanjing, China) was used to select
the stable clones which were transfected with pcDNA3.1-PGK1.
Establishment of the nude mouse tumor
xenograft models
Male Balb/c nude mice, 6–8 weeks old, purchased from
the Model Animal Research Center of Nanjing University, were used
in the experiments. Mice were maintained under specific
pathogen-free conditions at a constant room temperature and
humidity and a 12-h light/dark cycle. Sterilized food and water
were provided ad libitum. Animals were subjected to an
adaptation period of 2 weeks before the experiments. Transplantable
U251 cells, RR-U251 cells and cells stably treated with
pcDNA3.1-PGK1 in log phase growth were collected. Then, the mice
received subcutaneous (s.c.) implantation of the cells
(3×107) which were diluted in 100 μl phosphate-buffered
saline (PBS) containing 50% Matrigel (BD Biosciences, Franklin
Lakes, NJ, USA) by using 0.1 ml/inoculation in the lower back
region of the mice.
Statement of ethics
Animal studies were carried out in accordance with
the US National Institutes of Health Guidelines and followed the
rules of the National Animal Protection of China. The present study
was approved by the Medical Ethics Committee of Nanjing Brain
Hospital Affiliated to Nanjing Medical University (ethic no.
2011KY001). All efforts were made to minimize suffering and to
reduce the number of mice used.
Delivery of shRNA in vivo and
radiotherapy
When the s.c. tumors of the nude mice were ~60–80
mm3 in size, shRNA-PGK1 (500 pmol/tumor, 25 μl injection
volume) was slowly injected into the tumors at different sites.
Then, the tumors were immediately electrically pulsed (electric
field strength, 180 V/cm; pulse duration, 20 msec; pulse number, 6;
frequency of pulses, 1 Hz) using Square Wave pulse (Ningbo Scientz
Biotechnology, Ningbo, China) though 2 parallel stainless steel
electrodes on each side of the long diameter of the tumor in
situ. The negative control group was injected with shRNA-NC and
the control group with PBS. Fifty-four nude mice were divided
randomly and equally into 9 groups, and the groups were as follows:
i) U251+shRNA-PGK1; ii) U251+shNC; iii) U251+PBS; iv)
RR-U251+shRNA-PGK1; v) RR-U251+shNC; vi) RR-U251+PBS; vii)
overexpressed-PGK1 U251+shRNA-PGK1; viii) overexpressed-PGK1
U251+shNC; and viiii) overexpressed-PGK1 U251+PBS. Groups were
administered the indicated treatment every 24 h for 14 days. After
the first treatment, groups were treated with a dose of 5 Gy of
radiation by a 60Co source at 0.5 Gy/min for 10 min
every 3 days until the cumulative dose of radiotherapy reached 20
Gy. The tumor size was measured every other day with calipers, and
the tumor volume (V) was measured with all measurements in
millimeters using the following formula: V = 0.523 × length ×
width2.
Real-time polymerase chain reaction
(RT-PCR)
Total RNA was extracted from an equal number of
cells in vitro and the tumor samples with the SV Total RNA
Isolation System (Promega, Madison, WI, USA) according to the
manufacturer’s protocol and quantified by UV absorbance
spectroscopy. RNA was reverse transcribed into cDNA with a reverse
transcription system (Promega). mRNA expression was determined by
RT-PCR using GoTaq® qPCR Master Mix (Promega) under
standard thermocycler conditions (Eppendorf AG 22331, Germany). The
primers for human PGK1 were forward, 5′-ATGCTGAGGCTGTCACTCGG-3′ and
reverse, 5′-CACAGCAAGTGGCAGTGTCTCC-3′; and for GAPDH: forward,
5′-CGCTGAGTACGTCGTGGAGTC-3′ and reverse,
5′-GCTGATGATCTTGAGGCTGTTGTC-3′. The amplification process was 95°C
for 2 min, then 40 cycles at 95°C for 45 sec, 58°C for 45 sec, 72°C
for 45 sec, followed by 72°C for 10 min. The level of PGK1
expression in the different tumor groups was presented as the
theshold cycle value. The level of PGK1 mRNA was normalized against
the reference gene (GAPDH). Relative gene expression values were
measured using the 2−ΔΔCt method as following: Ratio =
2−ΔΔCt where ΔCt = CtPGK1 -
CtGAPDH, ΔΔCt = ΔCtexperimental group -
ΔCtcontrol group.
Western blot analysis
Total protein was extracted from an equal number of
cells in each group and an equal weight of tumor in each group with
a mixture of RIPA lysis buffer (Thermo Scientific, Runcorn UK) for
30 min. Following centrifugation at 12,000 × g for 10 min at 4°C,
the supernatant was collected. All samples were diluted in loading
buffer (Sunshine Biotechnology, Nanjing, China) and boiled for 3
min. Total protein (30 μg) was separated on 10% sodium dodecyl
sulfate-polyacrylamide gel by electrophoresis (SDS-PAGE). The
fractionated proteins were electro-transferred to a PVDF membrane
(Sunshine Biotechnology). The membrane was blocked in 5% skim milk
and probed with antibodies from mouse against human PGK1 (Abcam,
Cambridge, MA, USA) diluted in TBST buffer (1:1,000) overnight at
4°C. Then the membrane was incubated in the goat anti-mouse
horseradish peroxidase (HP)-conjugated secondary antibody
(1:10,000) for 1 h at room temperature. Immunoreactive bands were
detected using ECL chemiluminescence reagent (Thermo Scientific)
and visualized by the gel image analysis software.
Immunohistochemistry
Once the cumulative radiation dose reached 20 Gy,
mice were euthanized and tumor issues were quickly excised. The
tumors were fixed in neutral formalin solution and 4-mm sections
were cut from each paraffin block. The sections were dewaxed in
xylene and rehydrated, and an antigen retrieval step was carried
out. Then, the sections were incubated in 3%
H2O2 in PBS for 30 min, and blocked in PBS
containing 3% normal goat serum, 0.3% (w/v) Triton X-100 and 0.1%
BSA (room temperature for 1 h), followed by incubation with the
primary antibody mouse against human PGK1 (1:1,000) at 4°C
overnight. Subsequently, sections were developed with the ABC kit
and detected by DAB (both from Vector Laboratories, Burlingame, CA,
USA). The sections were then counterstained with hematoxylin,
dehydrated and mounted. The images were analyzed by Image-Pro
Plus.
Statistical analysis
The analysis was performed using SPSS 13.0.
Statistical significance was analyzed by the Student’s t-test and
the Mann-Whitney U test. Data are expressed as the means ± standard
deviation (SD). p<0.05 was considered to indicate a
statistically significant difference.
Results
Overexpression of PGK1 in human glioma
cells
U251 cells were stable transfected with the
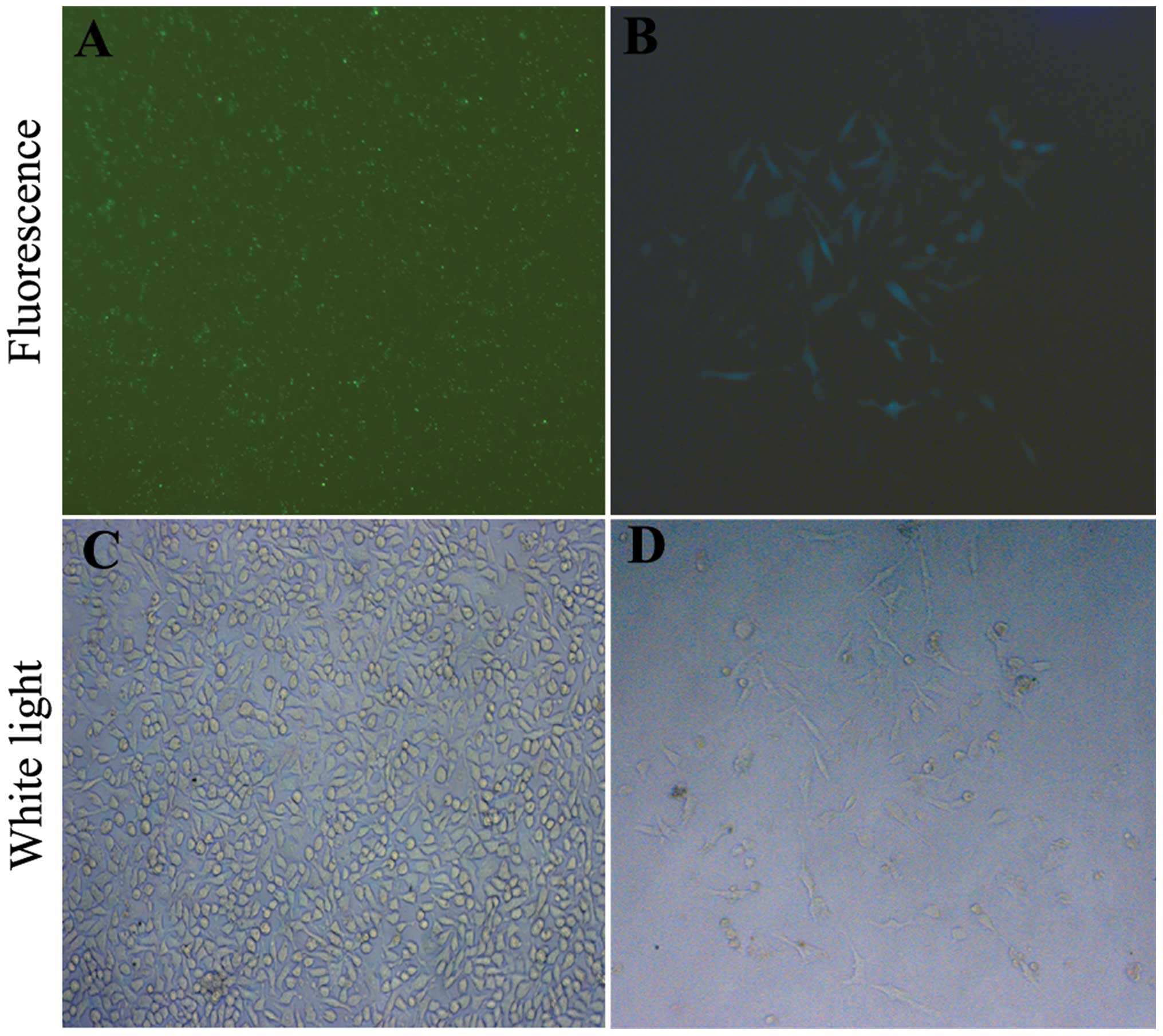
pcDNA3.1-PGK1 plasmid, and visible green fluorescence was observed
by fluorescence microscopy after transfection (Fig. 1A and C). The efficiency was ~85–90%.
Two weeks after the screening, most cells were killed by G418 (400
μg/ml); only a small number of cells survived and formed clones
(Fig. 1B and D). Stable clones were
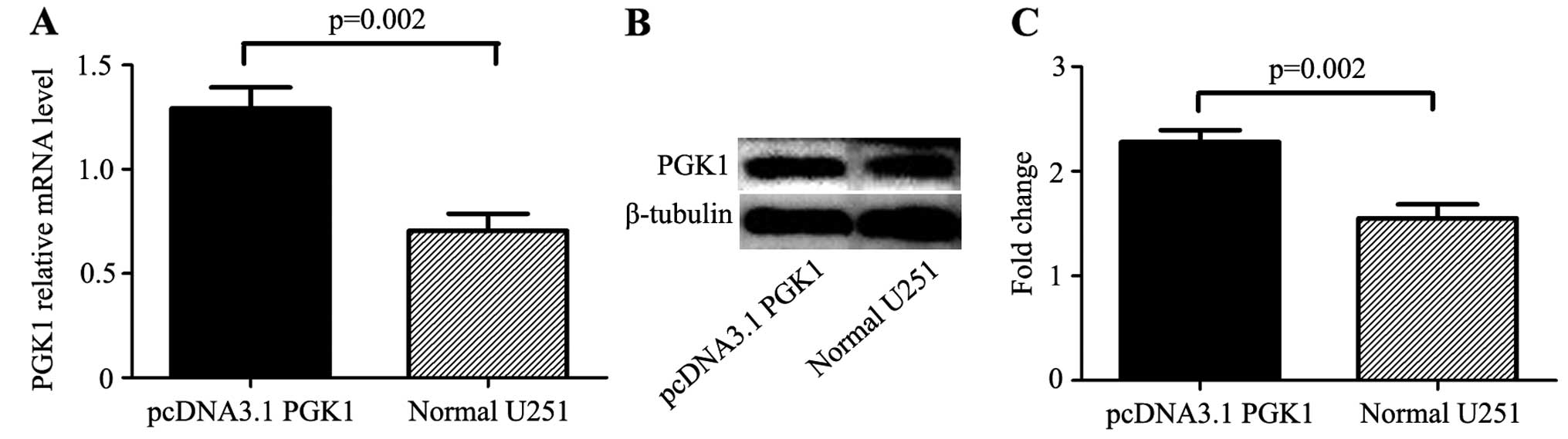
selected after 3 weeks. The mRNA and protein levels of PGK1 were
upregulated in the U251 cells treated with pcDNA3.1-PGK1 as
determined by real-time PCR and western blotting (Fig. 2). The expression of PGK1 was
significantly higher in the pcDNA3.1-PGK1 group when compared with
the expression in the control group at the mRNA level (1.29±0.08
vs. 0.71±0.07, p=0.02) and at the protein level (2.28±0.09 vs.
1.55±0.11, p=0.02).
Selection of the most efficient shRNA
specific to PGK1
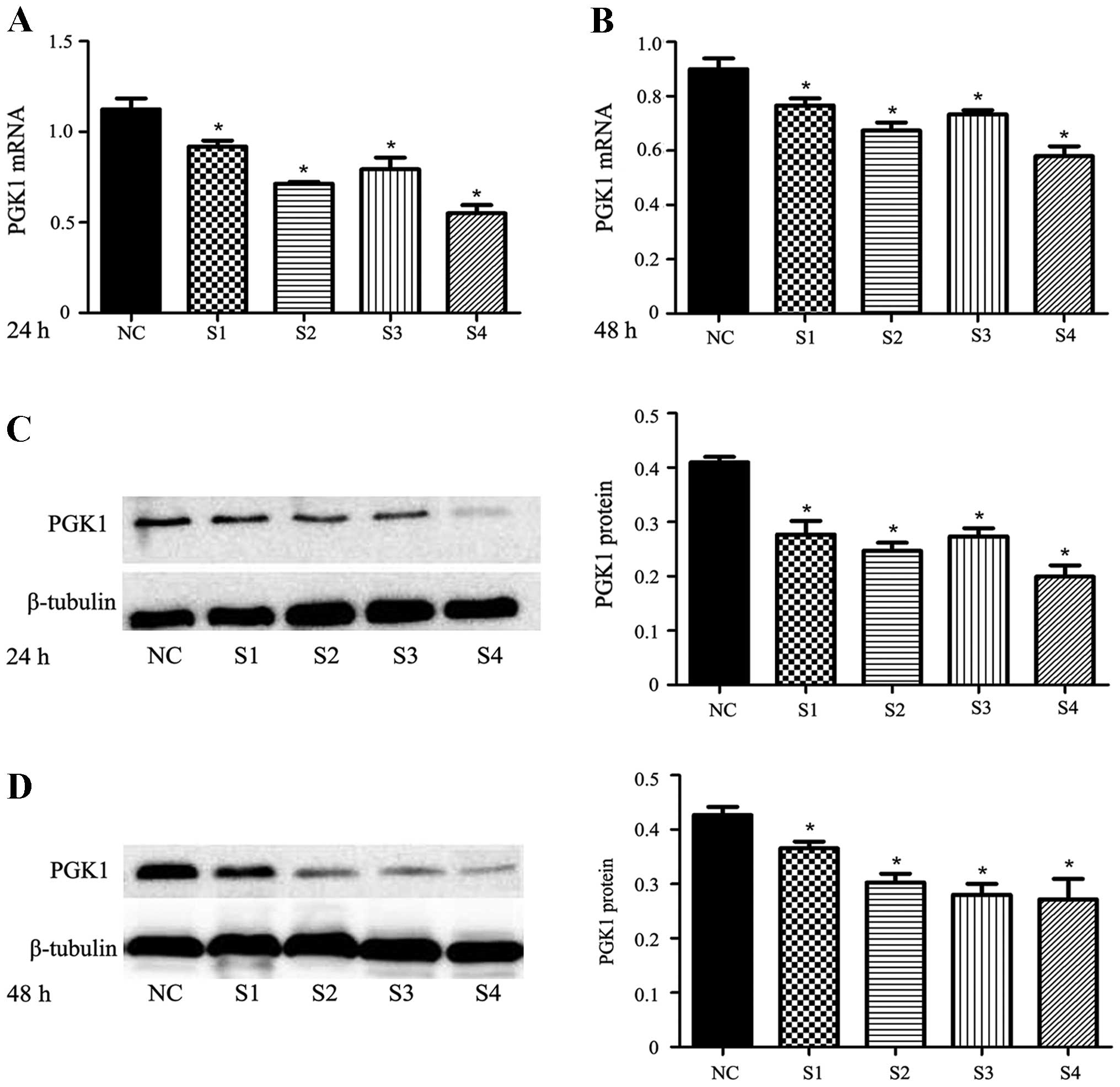
Transfection of shRNA-PGK1 duplexes led to a stable
exogenous gene expression with ~85–90% efficiency in the U251
cells; all four duplexes showed an inhibitory effect on PGK1
expression at the mRNA and protein levels (p<0.05) (Fig. 3). In order to select the most
efficient shRNA and the appropriate time point at which to
effectively inhibit PGK1, we further analyzed the expression of
PGK1 at the mRNA and protein levels at 24 and 48 h after
transfection. Through pairwise comparison, we found that the
expression of PGK1 inhibited by shRNA4 was significantly lower than
that by the other 3 series. The inhibition ratio of PGK1 expression
was 50.80±5.12% at the 24-h time point and 35.35±5.62% at the 48-h
time point at the mRNA level (p<0.05). The inhibition ratio of
PGK1 expression was 51.32±4.70% at the 24-h time point and
36.56±5.48% at the 48-h time point at the protein level
(p<0.05). The results showed that shRNA4 was more efficient in
inhibiting PGK1 expression compared with shRNA1, shRNA2 and shRNA3
at the 24-h time point after transfection. Based on the results,
shRNA4 transfection for 24 h was used for the following experiments
aimed at determining whether shRNA enhances the radioresistance of
PGK1 tumor xenografts.
Combination of shRNA-PGK1 with
radiotherapy in the U251 xenografts
To investigate the functional role of PGK1 in glioma
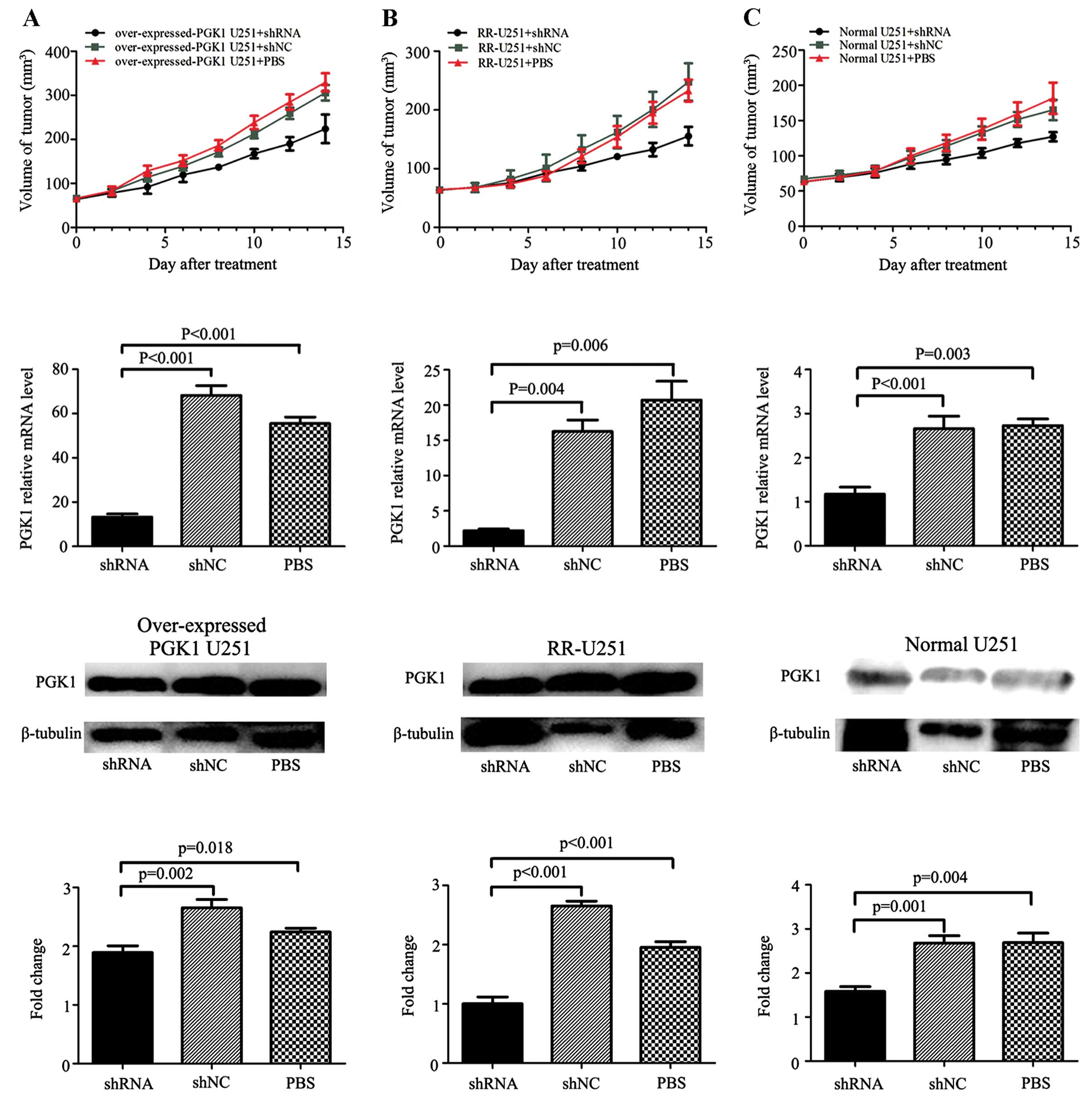
growth in vivo, tumors were measured every other day. The
growth of xenografts was inhibited following treatment with
PGK1-shRNA in the normal U251 group, RR-U251 group and
overexpressed-PGK1 U251 group. Approximately 2 days after the first
treatment, overexpressed-PGK1 U251 xenografts acquired a faster
growth rate compared with the normal U251 and RR-U251 group tumors
(Fig. 4). At day 14 post-treatment,
regarding the PBS-treated groups, the tumor size in the RR-U251
group was much larger than that in the normal U251 group
(232.9±18.6 vs. 181.7±22.2 mm3, p=0.002), and the tumor
size in the overexpressed-PGK1 U251 group was also larger than that
in the normal U251 group (329.6±20.6 vs. 181.7±22.2 mm3,
p=0.000). Combination of shRNA-PGK1 and radiotherapy significantly
decreased the tumor size in the normal U251 xenografts (shRNA-PGK1
group vs. shRNA-NC group: 127.2±6.0 vs. 165.0±13.2 mm3,
p=0.001), and following combination with shRNA-PGK1, the tumor size
also decreased comparing with the control group both in the RR-U251
xenografts (shRNA-PGK1 vs. shRNA-NC group: 155.4±14.5 vs.
247.7±29.0 mm3, p=0.000) and overexpressed-PGK1 U251
xenografts (shRNA-PGK1 vs. shRNA-NC group: 223.9±29.7 vs.
305.9±16.1 mm3, p=0.001). The data indicate that high
expression of PGK1 is correlated with enhanced ability of tumor
xenograft radioresistance, and shRNA-PGK1 effectively enhanced the
radiosensitivity of the U251 xenografts.
Furthermore, we assessed the quantity (means ± SD)
of PGK1 expression by RT-PCR and western blotting as described in
Materials and methods (Fig. 4). At
the mRNA level, the data for the group treated by shRNA vs. shNC
and PBS in the overexpressed-PGK1 U251 groups were respectively
13.25±1.15, 68.21±3.60 and 55.53±2.39; the data for the RR-U251
groups were 2.19±0.94, 16.25±1.33 and 20.72±2.17; the data for the
normal U251 groups were 1.17±0.13, 2.66±0.23 and 2.732±0.12 (all
p<0.05). While at the protein level, the data for the group
treated by shRNA vs. shNC and PBS in the overexpressed-PGK1 U251
groups were respectively 1.89±0.09, 2.65±0.11 and 2.24±0.05; the
data for the RR-U251 groups were 1.00±0.09, 2.65±0.06 and
1.95±0.08; the data for the normal U251 groups were 1.58±0.09,
2.68±0.14 and 2.69±0.18 (all p<0.05). Visually, xenograft tumor
sections showed extensive PGK1 staining by immunohistochemistry
(Fig. 5), confirming that shRNA
downregulated PGK1 expression in the xenografts in all groups. As a
result, shRNA-PGK1 significantly downregulated PGK1 in the U251
xenografts warranting additional investigation of its therapeutic
potential in glioma in the clinic.
Discussion
One important multimodal treatment of gliomas,
surgical removal of the tumor followed by radiotherapy, has
improved the survival of glioma patients. However, the frequency of
recurrence and rapid progression in patients emphasize the need for
a major enhancement of therapeutic efficacy to achieve long-term
survival without relying exclusively on uncertain resistance to
radiotherapy. In our experiment, we evaluated the expression of
PGK1 in specimens from different models of nude mouse tumor
xenografts. To our knowledge, this is the first in vivo
study that has shown the relationship between PGK1 regulation by
shRNA and U251 xenograft radiosensitivity. Tumors with high PGK1
expression showed higher tumorigenic properties which is consistent
with our previous study (18).
Notably, downregulation of the expression of PGK1 by specific small
molecule inhibitors can significantly decrease the radioresistance
and the progression of glioma.
It is important to note that cancer is not only a
genetic disease, but is also a type of energy metabolic disease
(19). Warburg proposed that the
primary cause of cancer is an energy deficiency even in the
presence of a high O2 concentration caused by an
irreversible damage to the mitochondrial function that induces
increased glycolysis which has been found in many cancers by
fluorodeoxyglucose positron emission tomography (20,21).
Despite the universality of PET, the study of the ‘bioenergetic’
basis behind this phenotype has not been examined clearly,
frequently or commonly by the scientific community during the past
several decades. As known, hypoxia mediated by the Warburg effect
is an intrinsic factor to subsequent tumor development and is a
prognostic variable for unfavorable outcome, as it provides the
mechanisms by which tumors can selectively promote a more
aggressive phenotype, recruit a nutritional supply, and provide
essential metabolic adaptations to ensure survival. The tumor
hypoxic microenvironment can trigger the altered metabolism of the
molecular basis of malignant gliomas: the glycolytic enzymes. PGK1,
as a vital glycolytic enzyme, plays a vital role and catalyzes the
transfer of a high-energy phosphoryl group from the acyl phosphate
of 1,3-diphosphoglycerate to ADP to produce ATP. In addition to our
previous study (18), Zhang et
al also found that overexpression of PGK1 is related to
increased necessity of energy in fast growing tumors due to protein
synthesis and degradation pathways (22). In the microenvironment, hypoxia
augments the expression of key mediators, such as HIF-1, c-myc and
MYCN, which have been related to PGK1 and resistance to
radiotherapy (23–26). These factors can hasten the
expression and the activity of PGK1. For example, PGK1 was induced
by one of the above factors, HIF-1, a key transcription factor
involved in glycolytic energy metabolism; its expression is
correlated with treatment resistance and overall poor prognosis.
HIF-1 can also promote the production of vascular endothelial
growth factor (VEGF) which in turn stimulates both angiogenesis and
glycolytic enzyme activity with the ability to facilitate anaerobic
production of ATP (26). Notably, a
recent study also demonstrated that HIF-1 is not only induced by
hypoxia, but also by glycolytic metabolites (27). Thus, we hypothesized that the
combinatorial effects of PGK1 and HIF-1 contribute to the poor
sensitivity to radiotherapy. Downstream targets of PGK1, E-cadherin
and β-catenin (13), are reportedly
associated with tumorigenic properties, including cell viability,
migration, invasion and angiogenesis ability which further promote
tumor radioresistance. This is consistent with our data of U251
xenografts showing that PGK1 is essential for increased tumor size
and significant resistance to radiotherapy.
Furthermore, the accumulation of lactate which is
the final product of glycolysis, increased by PGK1, may also
facilitate radioresistance. Lactate production may be the result of
a compensatory increase in the constitutive level of expression of
glycolytic enzymes, leading to a high rate of glycolytic capacity
(28). Lactate is expected to lead
to acidosis of the cell culture medium during hypoxia, resulting in
glioma radioresistance. The hypothesis is consistent with a study
by Hsu and Sabatini (29) who found
that glycolysis pathway enzymes have an antagonized apoptotic
effect which can lead to the tolerance to radiotherapy on apoptosis
of malignant tumors. Moreover, lactic acid enables the tumor in an
acidic tumor microenvironment to favor glioma invasion obtaining
tumor immune escape by inhibiting lymphocyte function compared with
the edge of glioma cells which can have relatively high oxygen
content and make use of lactic acid for oxidative
phosphorylation.
Ionizing radiation results in tumor DNA damage,
mainly by double-strand breaks, making them unable to divide and
grow (30). As a result of the
glycolysis of PGK1, it further affects DNA replication responses
and the repair system (31–33). The present study indicated that
overexpression of PGK1 led to glioma radioresistance, which may
also be due to the DNA damage repair ability of PGK1 and can be a
serious hurdle to diminish susceptibility of the irradiated glioma
cells to undergo apoptosis. This finally leads to the occurrence of
tumor radioresistance and recurrence. Yet, additional studies are
needed to confirm this hypothesis with cell cycle checkpoint
pathways which have been proven to play an important role in the
development of radioresistance in tumor cells (34,35).
Overall, our results revealed that PGK1 is
sufficient to promote radioresistance, and appropriate shRNAs can
significantly downregulate PGK1 in U251 xenografts. This confirms
our previous study that PGK1 can enhance radioresistance in
clinical glioma and U251 cells in vitro. However, future
investigation into the regulation of the enzymatic activity of PGK1
in additional tumor cell lines is critical. Among these tumor cell
lines, neural stem cells (NSCs) have attracted more and more
attention. The cells can act though the preferential activation of
the DNA damage response, induction of cell cycle arrest and then
repair of damaged DNA to improve radiation resistance (36). Zieker et al (37) initially explained the relationship
between PGK1 and stem cells in gastric cancer. Ultimately, based on
a better understanding of the role of PGK1 in glioma progression
and post-operative resistance, pharmacological agents designed as
agonists of the silencing of PGK1 may slow tumor growth or may
convert a therapeutically resistant tumor to one that is sensitive
to treatment.
Acknowledgements
The present study was supported by the grant
NSFC81172390 from the National Science Foundation of China, and the
grant ZKX10021 from the Health Bureau of Nanjing.
References
|
1
|
De Witt Hamer PC, Robles SG, Zwinderman A,
Duffau H and Berger M: Impact of intraoperative stimulation brain
mapping on glioma surgery outcome: a meta-analysis. J Clin Oncol.
30:2559–2565. 2012.PubMed/NCBI
|
|
2
|
Smith JS, Chang EF, Lamborn KR, et al:
Role of extent of resection in the long-term outcome of low-grade
hemispheric gliomas. J Clin Oncol. 26:1338–1345. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sanai N and Berger MS: Glioma extent of
resection and its impact on patient outcome. Neurosurgery.
6:753–764. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Benjamin R, Capparella J and Brown A:
Classification of glioblastoma multiforme in adults by molecular
genetics. Cancer J. 9:82–90. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Behin A, Hoang-Xuan K, Carpentier AF and
Delattre JY: Primary brain tumours in adults. Lancet. 361:323–331.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Höckel M, Schlenger K, Mitze M, Schäffer U
and Vaupel P: Hypoxia and radiation response in human tumors. Semin
Radiat Oncol. 6:3–9. 1996.
|
|
8
|
Unruh A, Ressel A, Mohamed HG, et al: The
hypoxia-inducible factor-1α is a negative factor for tumor therapy.
Oncogene. 22:3213–3220. 2003.
|
|
9
|
Ahmad SS, Glatzle J, Bajaeifer K, et al:
Phosphoglycerate kinase 1 as a promoter of metastasis in colon
cancer. Int J Oncol. 43:586–590. 2013.PubMed/NCBI
|
|
10
|
Chen G, Gharib TG, Wang H, et al: Protein
profiles associated with survival in lung adenocarcinoma. Proc Natl
Acad Sci USA. 100:13537–13542. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zieker D, Königsrainer I, Tritschler I, et
al: Phosphoglycerate kinase 1 a promoting enzyme for peritoneal
dissemination in gastric cancer. Int J Cancer. 126:1513–1520.
2010.PubMed/NCBI
|
|
12
|
Wang J, Ying G, Wang J, et al:
Characterization of phosphoglycerate kinase-1 expression of stromal
cells derived from tumor microenvironment in prostate cancer
progression. Cancer Res. 70:471–480. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang J, Wang J, Dai J, et al: A glycolytic
mechanism regulating an angiogenic switch in prostate cancer.
Cancer Res. 67:149–159. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cecconi D, Palmieri M and Donadelli M:
Proteomics in pancreatic cancer research. Proteomics. 11:816–828.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lincet H, Guével B, Pineau C, et al:
Comparative 2D-DIGE proteomic analysis of ovarian carcinoma cells:
toward a reorientation of biosynthesis pathways associated with
acquired platinum resistance. J Proteomics. 75:1157–1169. 2012.
View Article : Google Scholar
|
|
16
|
Duan Z, Lamendola DE, Yusuf RZ, Penson RT,
Preffer FI and Seiden MV: Overexpression of human phosphoglycerate
kinase 1 (PGK1) induces a multidrug resistance phenotype.
Anticancer Res. 22:1933–1941. 2002.PubMed/NCBI
|
|
17
|
Yan H, Yang K, Xiao H, Zou YJ, Zhang WB
and Liu HY: Over-expression of cofilin-1 and phosphoglycerate
kinase 1 in astrocytomas involved in pathogenesis of
radioresistance. CNS Neurosci Ther. 18:729–736. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding H, Cheng YJ, Yan H, et al:
Phosphoglycerate kinase 1 promotes radioresistance in U251 human
glioma cells. Oncol Rep. 31:894–900. 2014.PubMed/NCBI
|
|
19
|
Wu W and Zhao S: Metabolic changes in
cancer: beyond the Warburg effect. Acta Biochim Biophys Sin.
45:18–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gatenby RA and Gillies RJ: Why do cancers
have high aerobic glycolysis? Nat Rev Cancer. 4:891–899. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Seemann MD: PET/CT: fundamental
principles. Eur J Med Res. 9:241–246. 2004.PubMed/NCBI
|
|
22
|
Zhang DH, Tai LK, Wong LL, Chiu LL, Sethi
SK and Koay ES: Proteomic study reveals that proteins involved in
metabolic and detoxification pathways are highly expressed in
HER-2/neu-positive breast cancer. Mol Cell Proteomics.
4:1686–1696. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Luo FM, Liu XJ, Yan NH, et al:
Hypoxia-inducible transcription factor-1α promotes hypoxia-induced
A549 apoptosis via a mechanism that involves the glycolysis
pathway. BMC Cancer. 6:262006.
|
|
24
|
Qing G, Skuli N, Mayes PA, et al:
Combinatorial regulation of neuroblastoma tumor progression by
N-Myc and hypoxia inducible factor HIF-1α. Cancer Res.
70:10351–10361. 2010.PubMed/NCBI
|
|
25
|
Aebersold DM, Burri P, Beer KT, et al:
Expression of hypoxia-inducible factor-1α: a novel predictive and
prognostic parameter in the radiotherapy of oropharyngeal cancer.
Cancer Res. 61:2911–2916. 2001.
|
|
26
|
Ryan HE, Lo J and Johnson RS: HIF-1 alpha
is required for solid tumor formation and embryonic
vascularization. EMBO J. 17:3005–3015. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lu H, Forbes RA and Verma A:
Hypoxia-inducible factor 1 activation by aerobic glycolysis
implicates the Warburg effect in carcinogenesis. J Biol Chem.
277:23111–23115. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rodríguez-Enríquez S and Moreno-Sánchez R:
Intermediary metabolism of fast-growth tumor cells. Arch Med Res.
29:1–12. 1998.
|
|
29
|
Hsu PP and Sabatini DM: Cancer cell
metabolism: Warburg and beyond. Cell. 134:703–707. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ward JF: Radiation mutagenesis: the
initial DNA lesions responsible. Radiat Res. 142:362–368. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
VandeBerg JL: The phosphoglycerate kinase
isozyme system in mammals: biochemical, genetic, developmental, and
evolutionary aspects. Isozymes Curr Top Biol Med Res. 12:133–187.
1985.PubMed/NCBI
|
|
32
|
Popanda O, Fox G and Thielmann HW:
Modulation of DNA polymerases α, δ and ɛ by
lactate dehydrogenase and 3-phosphoglycerate kinase. Biochim
Biophys Acta. 1397:102–117. 1998.PubMed/NCBI
|
|
33
|
Vishwanatha JK, Jindal HK and Davis RG:
The role of primer recognition proteins in DNA replication:
association with nuclear matrix in HeLa cells. J Cell Sci.
101:25–34. 1992.PubMed/NCBI
|
|
34
|
McCord AM, Jamal M, Williams ES,
Camphausen K and Tofilon PJ: CD133+ glioblastoma
stem-like cells are radiosensitive with a defective DNA damage
response compared with established cell lines. Clin Cancer Res.
15:5145–5153. 2009.
|
|
35
|
Narayana A, Gruber D, Kunnakkat S, et al:
A clinical trial of bevacizumab, temozolomide, and radiation for
newly diagnosed glioblastoma. J Neurosurg. 116:341–345. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bao S, Wu Q, McLendon RE, et al: Glioma
stem cells promote radioresistance by preferential activation of
the DNA damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zieker D, Bühler S, Ustündag Z, et al:
Induction of tumor stem cell differentiation - novel strategy to
overcome therapy resistance in gastric cancer. Langenbecks Arch
Surg. 398:603–608. 2013. View Article : Google Scholar : PubMed/NCBI
|