Introduction
In Europe, colorectal cancer (CRC) is responsible
for 13.6% of all cancers (1).
Despite continuing research, the five-year age-standardised
survival rate in the UK remains approximately 46% (CRUK). Present
treatment imparts considerable morbidity. Therefore, a number of
strategies are being pursued to reduce this disease burden. Of
these strategies, early detection (including enhanced population
screening and prophylactic surveillance in high risk groups) and
prevention are considered the most promising.
Abundant evidence indicates that non-steroidal
anti-inflammatory drugs (NSAIDs), including aspirin, are
anti-tumorigenic and can prevent CRC (2–6 and
refs. therein). For instance, in chemical and genetic animal
models, NSAIDs reduce the initiation and progression of CRC (or
aberrant crypt foci) (7–11). Recent meta-analysis of a large
number of patient studies indicates that treatment with daily
aspirin for 5 years or longer reduces the risk of developing
multiple cancer types, including CRC (12). Furthermore, in a randomised
controlled trial of carriers with Lynch syndrome (CAPP2 study),
ingestion of a 600 mg daily dose of aspirin for ~2 years was
capable of substantially reducing CRC incidence (13). Aspirin has also been proposed to
suppress metastasis (14), and
regular aspirin use after diagnosis of non-metastatic CRC is
associated with a reduced CRC-specific mortality, an association
that was strongest when the primary tumour overexpressed
cyclooxygenase-2 (COX-2) (15).
Despite these promising data, the use of aspirin for
chemopreventative purposes universally is restricted by the
potential gastropathy associated with long-term use (16). Thus, there is an overwhelming
rationale to identify aspirin-related compounds that retain the
specific toxicity to CRC but are safer and more effective than
aspirin itself.
A number of derivatives of the salicylate/aspirin
molecule have been tested for toxicity to cancer cells. For
example, 5-aminosalicylic acid (mesalazine) improves replication
fidelity in HCT-116 CRC cells (17,18).
Moreover, NCX-4040 (an NO releasing aspirin, 2-(acetyloxy)benzoic
acid 4-[(nitrooxy)methyl]phenyl ester) can also suppress
microsatellite instability (19)
and sensitise human colon cancer cells, cultured in vitro
and transplanted in vivo in mice, to oxaliplatin (20). Another nitro-derivative of aspirin,
NCX-4016 (2-(acetyloxy)benzoic acid 3-[(nitrooxy)methyl] phenyl
ester), inhibits epidermal growth factor signalling in human
ovarian carcinoma and inhibits tumour growth in ovarian cancer
xenografts (21). MDC-43, a
para-positional isomer of phosphoaspirin
(4-[(diethoxyphosphoryloxy)methyl] phenyl 2-acetoxybenzoate)
reportedly inhibited the growth of cells derived from colon, lung,
liver, pancreas and breast cancer (22).
The mechanism(s) by which NSAIDs protect against CRC
are a matter of debate. As NSAIDs inhibit cyclooxygenase activity
and prostaglandins have been implicated in neoplastic growth
(23), a significant focus of
research has understandably been into the development of agents
that can reduce specific COX activity (24,25).
However, there is a growing consensus that NSAIDs can exert their
anti-proliferative activities via COX-independent effects (26,27).
Other mechanisms that have been invoked for the anti-proliferative
effects of these agents include: inhibition of the transcription
factor NF-κB (28); activation of
the p38 MAP kinase pathway with subsequent cyclin D1 degradation
(29); downregulation of Bcl-2
expression (30); activation of p53
and p21 dependent on the ATM checkpoint kinase (31); upregulation of 15-lipoxygenase-1
(32); the induction of expression
of pro-apoptotic DNA repair proteins (33); inhibition of epidermal growth factor
activation (34,35) and a prophylactic action wherein
selection for microsatellite stability occurs (36). Studying variants of aspirin and
related compounds may not only reveal more active/specific
alternatives, but may also allow insight into the chemical
components of the agents that are associated with these differing
mechanistic activities.
We recently generated a panel of variants of
salicylate in order to identify molecules that show the specific
effects of aspirin against CRC, but with increased
anti-proliferative activity. Of the compounds tested, ‘diaspirins’
[e.g. bis(2-carboxyphenyl) succinate] were noted to be
significantly more toxic to CRC cells than aspirin yet appeared to
retain some specificity to CRC cells (37).
Here, we extended this investigation. We generated
novel compounds that are based on the ‘diaspirins’ and demonstrated
that these compounds had potent antitumour activity against CRC
cells in vitro and in vivo, in a well characterised
implantable mouse model of CRC. Investigation of the mechanism of
action of these agents revealed that both agents stimulated the
NF-κB pathway and repressed NF-κB-driven transcription. However, it
also suggested that the mechanism underlying this repression
differs between aspirin and the diaspirin compounds. These data
identify novel anticancer compounds and shed light on the
structure-activity relationships important for the pro-apoptotic
response to aspirin.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM) and fetal
bovine serum (FBS) were obtained from PAA (Somerset, UK) or
Sigma-Aldrich (Dorset, UK). Precision Plus Protein Colour standards
and nitrocellulose were from Bio-Rad. RPMI-1640 medium,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
dimethyl sulfoxide (DMSO) and propidium iodide were from
Sigma-Aldrich. All other reagents were obtained from Sigma-Aldrich
unless otherwise stated.
Compound synthesis
Diaspirin [bis(2-carboxyphenyl) succinate; PN508]
and fumaryl diaspirin [bis(2-carboxyphenyl) fumarate; PN517] were
prepared as previously described (37). Aspirin is commercially available as
acetylsalicylic acid, and the synthesis of the other compounds is
as described in supplementary information (provided upon request).
The compounds tested are shown in Fig.
1.
Preparation of PN511 and PN512
These were prepared using the method described for
PN508/PN517 (37) using sebacoyl
chloride and terephthaloyl chloride, respectively. Both were
recrystallised from 90/10 toluene ethanol mixture. PN511 is a white
solid [mpt 135–136°C; literature mpt 139–142°C [see e.g. (45)]; IR (cm−1): 2917, 2854,
1754, 1683, 1604]. PN512 is a white solid [mpt 295–300°C, IR
(cm−1): 1736, 1698, 1605]. The molar masses of the
products were confirmed as 442 and 406 respectively by titration
with standardised sodium hydroxide using the method described
previously (37). Samples submitted
for accurate mass spectrometry returned the following data: PN511
m/z (CI) 460.1964 (M + NH4+,
C24H26O8NH4 requires
460.1966); PN512 m/z (CI) 424.1025 (M + NH4+,
C22H14O8NH4 requires
424.1027).
Preparation of PN524 and PN525
PN524 was prepared by reaction of 3-bromobenzoyl
chloride with salicylic acid in the presence of 1 mole-equivalent
of pyridine. 3-Bromobenzoyl chloride is available commercially but
can also be produced from 3-bromobenzoic acid by the action of
excess thionyl chloride (SOCl2). Refluxing for 2–3 h
(with trapping of the evolved gases) until all of the solid
material had dissolved, followed by removal of any excess thionyl
chloride by distillation, gave an almost quantitative yield that
can be used immediately in the subsequent step. Salicylic acid (0.1
mol, 13.81 g) was dissolved in THF (50 ml) and stirred. Pyridine
(0.1 mol, 8.2 ml) was then added and the mixture was stirred
magnetically while 3-bromobenzoyl chloride (~13.25 ml) was added
slowly over 2–3 min. The temperature rose to 35–40°C during the
addition. Pyridinium chloride was precipitated and stirring was
continued for a further 12–24 h. Ice-cold water (200 ml) was then
added and the mixture was stirred thoroughly. The mixture was
extracted with diethyl ether (150 ml) by vigorous magnetic
stirring. The ether layer was then separated and extracted with 10%
Na2CO3 solution (150 ml) to remove the PN524
from unwanted by-products. The carbonate layer was then acidified
with concentrated HCl until a precipitate was seen (pH 2.0–3.0).
The precipitate was taken up in diethyl ether, separated and dried
with anhydrous MgSO4 before rotary evaporation of the
ether. A clean white solid (17.0 g, 53%) was obtained.
Recrystallisation was achieved from boiling hexane with some
acetone added, by cooling to −15°C for 24–36 h. White crystals were
obtained [mpt 140–142°C; IR (cm−1): 3068, 1738, 1682,
1605]. The molar mass of the product was confirmed as 321 by
titration with standardised sodium hydroxide using the method
described previously (37).
PN525 was prepared in an analogous way using
4-methylbenzoyl chloride in place of 3-bromobenzoyl chloride. The
product is a white solid and was recrystallized from toluene [mpt
148–150°C, IR (cm−1): 1734, 1674, 1605]. The molar mass
of the product was confirmed as 256 by titration with standardised
sodium hydroxide using the method described previously (37).
Preparation of PN528
Methyl salicylate (13.0 ml) was placed in a 100-ml
conical flask. Pyridine (8.1 ml) was then added and the mixture was
stirred magnetically while benzoyl chloride (11.6 ml) was added
slowly over 2–3 min. The temperature rose to 48–50°C during the
addition. Crystals of pyridinium chloride precipitated and stirring
was continued for a further 2 h. Hexane (50 ml) was then added and
the mixture was stirred thoroughly. The resulting heavy white
precipitate (~35 g) was filtered and allowed to air dry before
being shaken vigorously with water (100 ml) to dissolve the
pyridinium chloride by-product. The remaining solid was filtered
under vacuum and dried at 60°C. The yield was 15–16 g (~65%). This
crude product had a mpt of 82–85°C. The product was recrystallised
by dissolving in the minimum volume of hot acetone and then adding
hot hexane until the solution just remained clear. The solution was
then cooled to −15°C for a minimum of 24 h. A white solid [12.0 g,
mpt 84–86°C; IR (cm−1): 1737, 1715, 1605] was obtained.
The literature mpt is 83.8–84°C (51).
Preparation of PN529
Isopropyl salicylate was reacted with a slight
excess of 3-bromobenzoyl chloride in the presence of pyridine with
no additional solvent. Isopropyl salicylate is available
commercially but we also prepared small quantities from 2-propanol
and salicylic acid by refluxing them with a catalytic quantity of
concentrated H2SO4. A ratio of 25 g salicylic
acid to 100 ml 2-propanol was used with 1–2 ml of concentrated
H2SO4. After a minimum of 12 h refluxing, the
excess 2-propanol was removed by rotary evaporation. The residue of
oily solid was shaken with 10% NaHCO3, with more solid
bicarbonate added until all the unreacted salicylic acid had
dissolved. This mixture was then extracted well with diethyl ether
before separating the ether layer, drying over anhydrous
MgSO4 and removing the ether by rotary evaporation. A
low yield (3–5 g; 9–15%) of almost pure isopropyl salicylate (tlc)
was obtained and used without further purification. 3-Bromobenzoyl
chloride was obtained as described for the preparation of PN524
above. Isopropyl salicylate (1.90 g) was mixed with pyridine (1.6
ml) and was then added carefully to 3-bromobenzoyl chloride (2.38
g). An immediate reaction produced a near-solid product. The
reaction vessel was protected with a silica gel guard tube and left
at room temperature for 2–3 days to complete the reaction. Water
(20 ml) was added to the tube which was stoppered and shaken
vigorously. A sticky oil was released which was taken up in diethyl
ether, washed with 10% NaHCO3 (20 ml) and then water (20
ml). The ether layer was dried over anhydrous MgSO4 and
rotary evaporated to give a clear, pale yellow oil (3.4 g; 87%). A
solid product was obtained by dissolving the oil in a little warm
diethyl ether and then diluting with boiling hexane or light
petroleum ether until a cloudy solution was observed. Storing this
mixture at −15°C for 2–3 days gave white crystals [mpt 49–51°C, IR
(cm−1): 2979, 1735, 1703, 1606].
Cell culture
The human colorectal adenocarcinoma cell line SW480
(supplied by ECACC) was cultured in Leibovitz’s L-15 medium
containing 10% (v/v) FBS supplemented with 1%
L-glutamine-penicillin-streptomycin (Sigma-Aldrich). The human
colorectal adenocarcinoma cell lines LoVo and HCT-116, the human
breast carcinoma cell line MCF-7 and the human endothelial cell
line EA.hy926 were all kindly provided by Dr Wang (University of
Wolverhampton) and maintained in DMEM containing 10% (v/v) FBS
supplemented with 1% L-glutamine-penicillin-streptomycin
(Sigma-Aldrich). The A549 human lung adenocarcinoma cell line was
provided by Dr Safrany (University of Wolverhampton) and maintained
in RPMI-1640 medium supplemented with 10% FBS with 1%
L-glutamine-penicillin-streptomycin. In cytotoxicity experiments
for IC50 determinations, to minimize medium-specific
variation, cells were cultured and tested in DMEM containing 10%
(v/v) FBS supplemented with 1% L-glutamine-penicillin-streptomycin.
The murine adenocarcinoma cell line MAC13 [supplied by Professor
Double, University of Bradford (38)] was maintained in RPMI-1640 medium
supplemented with 10% FBS/1% pen/strep solution/1% glutamine. The
SW480 cell line was maintained in L-15 medium, was cultured at 37°C
and regularly passaged at ~80% confluency. All other cells were
cultured at 37°C in a humidified incubator with 5% CO2
and regularly passaged at ~80% confluency.
Evaluation of the anti-proliferative
effect
Compounds were prepared as a stock (0.5 M) in DMSO,
prior to addition to cells. The cytotoxic effect of aspirin and the
other novel compounds was evaluated using the MTT assay (39) with modification (40). Briefly, 104 cells/well
were cultured overnight in 96-well microtitre plates. Twenty-four
hours after the initial seeding, the culture medium was discarded,
and the cells were incubated with medium containing drugs at the
requisite concentration and incubated for the tested time at 37°C,
then replaced with medium containing 300 μl of 0.5 mg/ml MTT per
well and incubated for 3 h at 37°C, then replaced with 200 μl of
DMSO and incubated for 10 min at 37°C to develop a coloured
formazan complex. Absorbance was recorded at 540 nm using a visible
plate reader (Labsystems Multiskan MS). Viability is reported as
the percentage of treated cells relative to the cells in control
wells. Compound anti-proliferative activity, stated as
IC50 values, was determined by interpolation or using
non-linear regression analysis using GraphPad Prism Statistics
Software package (ver. 6.0; San Diego, CA, USA).
For the evaluation of toxicity (in vitro) to
the MAC13 cell line, when plated cells reached 90% confluency they
were treated with either PBS, aspirin, DiA or F-DiA dissolved in
PBS. After 48 h at 37°C the cells were washed in PBS twice before
0.5 ml trypsin-EDTA (10%) was added, and the cell numbers were
determined using a Coulter Counter.
Preparation of drugs
For evaluation of the activity described in Figs. 3–5,
the compounds were prepared as a 1 M stock solution in DMSO and
sonicated to achieve solubility. DMSO concentrations never exceeded
0.5% of the medium volume. The medium containing drugs was
sonicated until all visible particles were dissolved. The pH was
subsequently adjusted to 7.6 with NaOH. Aspirin (Sigma, USA) was
solubilized in water using 10 M NaOH and the pH was adjusted to
7.0.
Apoptosis assays
Staining for cell surface phosphatidylserine
residues was carried out using an Annexin V -FITC apoptosis
detection kit (Oncogene Research Products) according to the
manufacturer’s instructions. The percentage of apoptotic cells was
measured using BD FACS Aria II (BD Biosciences, Franklin Lakes, NJ,
USA). A negative control without the Annexin V -FITC antibody and
DAPI were used to assess the background signal.
Cell cycle analysis
After drug treatment, SW480 cells were trypsinized
and pelleted for 5 min at 1,600 rpm. The supernatant was removed.
The cells were suspended in 100 μl citrate buffer and 450 μl of
solution A [10 mg trypsin type IX-S in 500 ml stock solution (2 g
trisodium citrate, 121 mg Tris-base, 1044 mg spermine
tetrahydrochloride, 2 ml Nonidet NP-40 in 2l dH2O, pH
7.6)] was added to the samples and vortexed for 2 min.
Subsequently, 375 μl solution B (250 mg trypsin inhibitor, 50 mg
RNAse A in 500 ml stock solution, pH 7.6) was added to the samples,
vortexed briefly and incubated for 10 min at room temperature.
Next, 250 μl solution C (208 mg propidium iodide, 500 mg spermine
tetrahydrochloride in 500 ml stock solution, pH 7.6) was added to
the samples and vortexed briefly and incubated on ice in the dark
for 10 min. Distribution of cells in the different phases of the
cell cycle was analysed using BD FACS Aria II.
Transfections and reporter assays
For the analysis of NF-κB-driven transcription,
cells were transfected with the 3′ enhancer CONA (3x κB ConA-Luc)
(supplied by Professor R.T. Hay, University of Dundee) and
pCMV-β-galactosidase (Promega) plasmids as described previously
(41). The relative luciferase
activity was calculated as units of luciferase activity per unit of
β-galactosidase activity.
Western blot analysis
Following treatment, cytoplasmic and nuclear
extracts were prepared as previously described (42). The protein concentration was
determined using a Bradford assay (Bio-Rad). Protein extracts
(20–50 μg) were resolved using discontinuous SDS-PAGE with a 10%
gel, and immunoblotting was conducted using standard procedures.
Primary antibodies used were: rabbit anti-cyclin D1 (Thermo
Scientific, USA; 1:1000), sheep anti-IκB (a kind gift from R.T.
Hay) and mouse anti-actin as a loading control. Proteins were
detected using western blotting luminol reagent (Santa Cruz, USA).
ImageJ software (NIH) was employed for densitometric analysis of
the western blots.
Implantable mouse CRC model
NMRI nude mice from the Aston colony were treated
with MAC13 cells (5×106) sub-cut into their flanks. Once
the tumour was established, it was dissected and small fragments
were re-transplanted (subcutaneously) into more mice. Mice (~20 g
in weight) were selected at random and administered PBS or DiA or
F-DiA, dissolved in PBS (1 mg/kg) intravenously. Animals were
randomly divided (6 animals/group). Medication was administered
intravenously daily. The mice were monitored daily for weight, food
and water consumption. Tumour size was recorded using callipers.
Prior to the animal model experiment, as much as 5 mg/kg compound
was given to several control mice and no toxicity was observed
(data available upon request). The experiment ended when the
control mouse tumours started to ulcerate. All animal experiments
were conducted under Home Office Licence in accordance with the UK
Animals (Scientific Procedures) Act 1986.
Statistical analysis
Values are expressed as means ± SD or SEM. To
evaluate the difference in means between groups in the animal
study, one-way ANOVA followed by Tukey’s post-hoc test was
undertaken.
Results
Aspirin analogues demonstrate potent
activity against CRC cells
To understand the molecular basis for the antitumour
effects of aspirin and identify more effective alternatives, we
previously synthesised a series of derivatives of the aspirin
molecule. These studies revealed that diaspirin (DiA) and fumaryl
diaspirin (F-DiA) inhibit proliferation of CRC cell lines at
significantly lower concentrations than aspirin (37). To extend these studies and identify
further lead molecules, we synthesised an additional series of
aspirin derivatives (Fig. 1).
Cytotoxicity (MTT) assays demonstrated that at 0.5 mM
(pharmacologically relevant dose for aspirin), DiA (PN508) and
F-DiA (PN517) and to an even greater extent, isopropyl
m-bromobenzoylsalicylate (PN529) reduced the viability of SW480 CRC
cells (Fig. 2A). To investigate the
specificity of compound toxicity in more detail, we tested the
capacity of DiA, F-DiA and PN529 to affect proliferation of a
number of established cell lines in vitro (Fig. 2B and Table I), controlling for any variability
that could arise from cell culture conditions through culturing all
cells with DMEM as the basal medium. While we noted that culturing
SW480 cells in their non-native medium (DMEM rather than L-15
medium) reduced the sensitivity of the cells to the compounds
tested, DiA and F-DiA in this assay system arguably showed a
modicum of specificity towards the other CRC cell lines tested
(HCT116 and LoVo), and given our finding that PN529 can induce
necrosis (37), we focused our
attention on the anti-proliferative activity of DiA and F-DiA in
more detail in vitro and in vivo.
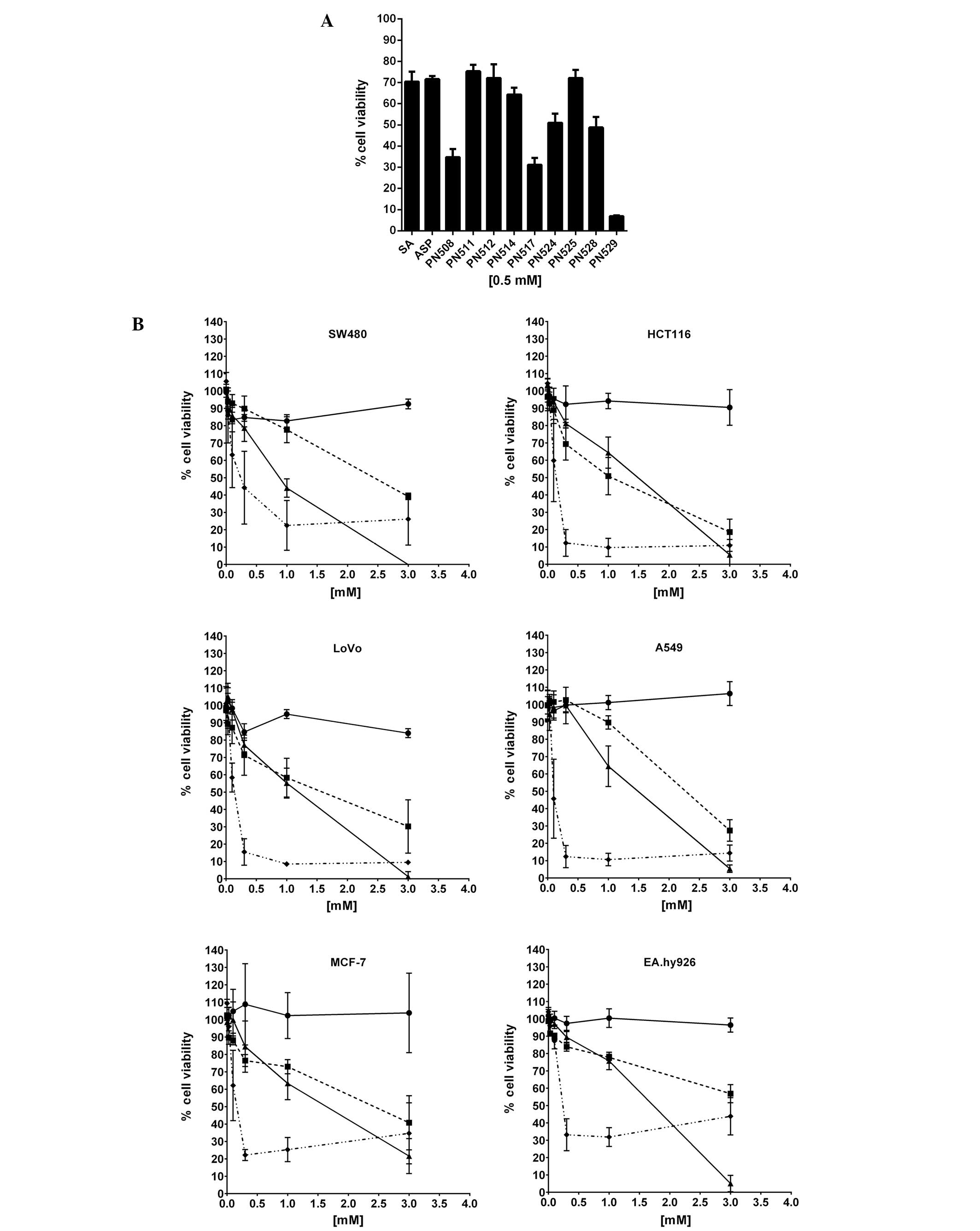 | Figure 2Cytotoxicity of aspirin analogues to
selected cancer cell lines in vitro. (A) SW480 cells were
plated at a density of 104 cells/ml in 96-well
microtitre plates (200 μl/well) in a L-15-based medium. Twenty-four
hours after seeding, the cells were treated with
compound-containing culture medium for a further 72 h. The
anti-proliferative effects were measured in an MTT assay. All
compounds were dissolved in DMSO. Mean ± SEM is shown (N=3). The
effect of salicylic acid (SA) and aspirin (ASP) is included for
comparison. (B) Sensitivity of selected colorectal (SW480, HCT116,
LoVo) and non-colorectal (A549, MCF7, EA.hy926) cancer cell lines
cultured in DMEM-based culture medium, to diaspirin (PN508),
fumaryl diaspirin (PN517) and isopropyl m-bromobenzoyl salicylate
(PN529). Twenty-four hours after seeding, the cells were treated
with compound-containing culture medium for a further 72 h. The
anti-proliferative effects were measured in an MTT assay. All
compounds were dissolved in DMSO. Each point represents the mean ±
SD (N=3). Circle, carrier molecule (DMSO); square, PN508 (DiA);
triangle, PN517 (F-DiA); diamond, PN529. |
 | Table IIC50 values (mM) for
compounds using the MTT assay to selected colorectal and
non-colorectal cancer cell lines. |
Table I
IC50 values (mM) for
compounds using the MTT assay to selected colorectal and
non-colorectal cancer cell lines.
| Compound (mM) |
|---|
|
|
|---|
| PN508 | PN517 | PN529 |
|---|
| Cell lines |
| SW480 | 2.43±0.1 | 0.87±0.05 | 0.24±0.06 |
| HCT116 | 1.08±0.2 | 1.47±0.1 | 0.15±0.01 |
| LoVo | 1.59±0.2 | 1.17±0.08 | 0.14±0.01 |
| A549 | 2.3±0.1 | 1.47±0.08 | 0.13±0.001 |
| MCF-7 | 2.4±0.2 | 1.62±0.2 | 0.15±0.04 |
| EA.hy926 | 6.2a | 1.68±0.09 | 0.24±0.2 |
The major anti-proliferative effect of aspirin is
recognised to be the induction of apoptosis. However, the mechanism
by which DiA and F-DiA act against CRC cells is as yet unknown. To
further understand this mechanism, we used Annexin V assays to
investigate the effects of aspirin derivatives on apoptotic cell
death. We found that both compounds (3 mM) induced a significant
increase in the percentage of SW480 CRC cells undergoing apoptosis,
compared to the carrier (DMSO)-treated controls (Fig. 3). This increase in apoptosis was
paralleled by a decrease in the percentage of viable cells,
confirming the death-inducing capacities of the agents (data not
shown). Time course studies revealed that 3 mM F-DiA mediated a
significant increase in apoptosis within 3 h of treatment (Fig. 3), while a more prolonged (>5 h)
exposure was required before DiA-mediated apoptosis was evident
(data not shown). In contrast to the diaspirin compounds, aspirin
at 3 mM had minimal effect on cell growth/death after an 18-h
exposure (data not shown), which is in keeping with previous
studies showing that 5 mM aspirin is required to induce detectable
apoptosis of these cells within this time frame (41). Taken together, these data indicate
that the diaspirin compounds induce apoptosis of CRC cells and that
this occurs at a lower concentration than for aspirin and that
F-DiA is the more active of the compounds.
Aspirin analogues induce the degradation
of cyclin D1, but do not affect cell cycle distribution
Having established that the aspirin analogues induce
apoptosis, we next aimed to determine whether the underlying
mechanism is similar to that of aspirin. Understanding the
mechanism of action of these agents may not only identify novel
apoptotic pathways, but may also reveal structural aspects of
aspirin that are important for its antitumour activity. Degradation
of cyclin D1 is an early response to aspirin and is critical for
its apoptotic activity (29).
Therefore, we firstly examined the effects of the agents on
cellular levels of this protein. We found that both DiA (3 mM, 18
h) and F-DiA (3 mM, 18 h) induced a marked decrease in cyclin D1
levels (Fig. 4A). Furthermore, this
decrease was time-dependent and for both agents, preceded the
induction of apoptosis (Fig. 4B).
However, unlike the G2/M cell cycle arrest observed in response to
aspirin, DiA- and F-DiA-mediated depletion of cyclin D1 was not
associated with significant changes in the cell cycle profile
(Fig. 4C).
Aspirin analogues stimulate the NF-κB
pathway
NF-κB is a ubiquitously expressed transcription
factor that plays a critical role in cell growth and death. It
generally resides in the cytoplasm as a heterodimer of the RelA/P50
subunits, bound to the inhibitor protein, IκB. In response to a
variety of agents, IκB is degraded allowing NF-κB to translocate to
the nucleus and regulate expression of target genes. Thoms et
al previously demonstrated that aspirin-induced degradation of
cyclin D1 is linked to the induction of apoptosis through
stimulation of this pathway (29).
Moreover, cyclin D1 is transcriptionally activated by NF-κB. To
determine whether DiA- and F-DiA-mediated depletion of cyclin D1
and induction of apoptosis are also associated with modulation of
NF-κB signalling, we firstly examined the effects of these agents
on IκBα. Fig. 5A demonstrates that
both DiA and F-DiA induced a marked decrease in cytoplasmic levels
of this protein, suggestive of proteasome-mediated degradation.
Following aspirin-mediated degradation of IκB, RelA
translocates from the cytoplasm to the nucleoplasm and then to a
nuclear compartment, the nucleolus (41,43).
This nucleolar compartmentalisation of RelA causes a reduction in
basal levels of NF-κB transcriptional activity, which is critical
for the apoptotic effects of the agent. Immunocytochemical analysis
demonstrated that DiA and F-DiA induced a substantial increase in
nucleoplasmic RelA, confirming that these agents stimulated the
NF-κB pathway. However, in contrast to aspirin, the protein did not
accumulate specifically in nucleoli (Fig. 5B and C). Time-course studies
demonstrated that for both compounds, translocation of RelA to the
nucleoplasm was subsequent to degradation of cyclin D1, but
preceded the induction of apoptosis (Fig. 5C). They also demonstrated that
F-DiA-mediated nucleoplasmic accumulation of RelA was more rapid
and significant than that induced by DiA, in keeping with the
apoptotic effects of the agents. Indeed, nucleoplasmic RelA was
evident within 1 h of F-DiA treatment and cells appeared dead
within 3 h.
In response to specific stresses, nucleoplasmic
NF-κB/RelA complexes have been shown to mediate apoptosis by
repressing basal NF-κB-driven transcription (44). To examine this as a possible
mechanism for the apoptotic effects of the diaspirin compounds, we
next performed NF-κB reporter assays. We found that both DiA and
F-DiA induced a significant decrease in basal levels of NF-κB
transcriptional activity. This decrease was more pronounced than
for aspirin, occurred in a time-dependent manner and was subsequent
to nucleoplasmic accumulation of RelA (Fig. 5C and D). Furthermore, similar to
depletion of cyclin D1, nucleoplasmic accumulation of RelA and the
induction of apoptosis, we found that F-DiA had a more rapid and
profound effect on NF-κB-driven transcription than DiA (Fig. 5E). These data suggest that, similar
to aspirin, the diaspirin compounds stimulate the NF-κB pathway and
mediate apoptosis through repression of NF-κB-driven transcription.
However, they also suggest that the mechanism underlying this
repression differs between aspirin and the related agents.
Inhibition of tumour growth by diaspirins
in an implantable CRC mouse model
Taken together, the above data suggest that the
diaspirin compounds, particularly F-Dia, have potent activity
against CRC cells. To determine whether these effects translate
into in vivo potency, we utilised the well characterised
MAC13 syngeneic mouse model of CRC (38). In the cytotoxicity assays MAC13
cells cultured in vitro were more sensitive to DiA and F-DiA
than aspirin (data not shown); in vivo, the mouse colon
carcinoma cells were implanted subcutaneously into NMRI mice and
treatment commenced 12 days after implantation. Low dose (1 mg/kg)
DiA or F-DiA was administered every day for 10 days by intravenous
injection. No overt side-effect was observed between the treatment
and vehicle-treated control group, and food and water intake was
unaffected. As shown in Fig. 6,
treatment with DiA and F-DiA significantly inhibited the growth of
the tumours, and almost complete suppression of growth was apparent
in the mice treated with F-DiA.
Discussion
Colon cancer is a major cause of cancer-related
mortality worldwide, particularly in Western societies. Present
therapies are associated with significant morbidity and generally
provide marginal benefit. Hence, there is an overwhelming rationale
for novel developments that would lead to a reduction in incidence
and/or in cancer-related morbidity and mortality. Herein, we
demonstrated that two analogues of aspirin had greater
anti-proliferative activity against CRC cells in vitro than
aspirin and salicylate and that these compounds exhibited a modicum
of specificity for CRC cell lines in vitro [Fig. 2, Table
I and ref. (37)]. Furthermore,
we also demonstrated that DiA and F-DiA had potent antitumour
activity in vivo, in an implantable mouse model of CRC
(Fig. 6) and showed no obvious
side-effects when introduced to mice intravenously over a 10-day
period. Taken together, these data identify DiA and F-DiA as
potential novel therapeutic agents for CRC. Although a collection
of related structures termed ‘diaspirins’ [in particular
bis(3,5-dibromosalicyl) fumarate] have long been known to cross
link haemoglobin (45), as far as
we are aware, this is the first report of the antitumour properties
of these agents.
The phenotypic studies indicated that the diaspirin
compounds were capable of inducing apoptosis (Fig. 3). On analysis of the mechanisms
underlying this apoptosis, we found that similar to aspirin, the
diaspirin compounds sequentially induced depletion of cyclin D1,
degradation of IκB, nuclear translocation of NF-κB/RelA complexes
and repression of NF-κB activity. However, in contrast to aspirin,
the aspirin analogues did not induce nucleolar translocation of
RelA or cell cycle arrest. Aspirin consists of acetyl and
salicylate moieties, both of which have their own individual
targets (46). The aspirin
analogues we generated retained the salicylate part of the molecule
but lost the acetyl component. Indeed, western blot analysis with
antibodies against acetylated lysine indicated that aspirin induced
a dose-dependent increase in protein acetylation, but this was not
observed in response to DiA and F-DiA (data not shown). Therefore,
one possibility is that the ability of aspirin to stimulate NF-κB
signalling is associated with the salicylate component of the
compound while nucleolar translocation of RelA and cell cycle
arrest are associated with its acetylating potential. In keeping
with this suggestion, salicylate has previously been shown to
activate p38 kinase activity (47),
which we have previously shown lies upstream of aspirin effects on
cyclin D1 and the NF-κB pathway (29).
Constitutive activity of NF-κB has been shown to
contribute to carcinogenesis in a number of cancer types, including
CRC (48). We previously
demonstrated that aspirin-mediated nucleolar sequestration of RelA
induces apoptosis, at least in part, by causing repression of this
basal NF-κB transcriptional activity (41). Here, we found that the aspirin
analogues also mediated a reduction in basal levels of NF-κB-driven
transcription. Furthermore, we found that this reduction preceded
the induction of apoptosis. The effects of the diaspirin compounds
on NF-κB transcriptional activity were more pronounced than for
aspirin, in keeping with the enhanced apoptotic activity of these
agents. Taken together, these data suggest that similar to aspirin,
the analogues mediate apoptosis by repressing NF-κB-driven
transcription. We note that the NSAID ibuprofen can induce IκBα
degradation, NF-κB nuclear localization and suppression of cyclin
D1 expression (49).
As outlined above, we did not observe nucleolar
sequestration of RelA in response to the diaspirin compounds,
suggesting that an alternative mechanism is responsible for the
effects of these agents on NF-κB transcriptional activity. Campbell
et al (44) previously
demonstrated that stimulation of the NF-κB pathway by specific
pro-apoptotic stimuli causes nucleoplasmic accumulation of
repressive RelA/NF-κB complexes that mediate apoptosis by
recruiting histone deacetylases (HDACs) to the chromatin of
NF-κB-regulated anti-apoptotic genes. We previously demonstrated
that aspirin induces the ubiquitination of RelA, which targets the
protein to the nucleolus (50).
Therefore, it may be that in the absence of the acetylating
component of aspirin, nucleoplasmic RelA cannot be ubiquitinated,
allowing the recruitment of alternative repressive factors such as
HDACs. Our finding that, of the two compounds, F-DiA had the most
rapid effect in all of the mechanistic assays and the most
significant activity against the growth of implanted tumour adds
weight to our suggestion that the antitumour activity of this agent
is caused by modulation of NF-κB signalling. It also identified
F-DiA as particularly significant for therapy development. From the
range of analogues studied to date it would seem that increased
activity is at least, in part, associated with the presence of two
(or perhaps more) suitably spaced aromatic rings and indicates the
significance of two salicylate moieties in the molecule separated
by ~8–10Å. We speculate that F-DiA may demonstrate greater toxicity
as a consequence of its greater symmetry and reduced number of
conformers.
To conclude, we identified diaspirins that have
potent anti-proliferative activity against CRC cells in
vitro and in vivo. The in vitro mechanistic
studies provide compelling evidence that these agents exert their
anti-proliferative effects against SW480 cells by causing
repression of NF-κB-driven transcription. The mechanistic studies
also helped to elucidate structure-function relationships for the
aspirin molecule. In particular, these findings provide insight
into the mechanisms of action of F-DiA and support further studies
into the potential of F-DiA as a chemotherapeutic agent. We wish to
test these agents in models of cachexia and in genetically defined
mouse models [e.g. Apc(Min/+) and Apc(Min/+), Msh2(−/−)] of CRC. We
acknowledge, however, that formal dose ranging and toxicity studies
with, for example, clinical scoring of gastrointestinal toxicity
does need to be undertaken.
Acknowledgements
The authors thank Keith Holding, Keith Thompson and
Elizabeth Freyer (UoE) for technical assistance, Craig Nicol (UoE)
for help with manuscript preparation and Paul Perry and Matthew
Pearson (UoE) for help with microscopy. We also thank Dr Paul
Hooley and Dr James Vickers for comments on the manuscript. This
project was supported by funding from the Research Institute in
Healthcare Science, University of Wolverhampton (IDN) and by the
AICR (10-0158) (LAS). We gratefully acknowledge the support of
Professor Ian Oakes, University of Wolverhampton.
Abbreviations:
|
ASA
|
acetylsalicylic acid
|
|
ASP
|
aspirin
|
|
CRC
|
colorectal cancer
|
|
DiA
|
diaspirin [bis(2-carboxyphenyl)
succinate]
|
|
COX
|
cyclooxygenase
|
|
F-DiA
|
fumaryl diaspirin
|
|
mpt
|
melting point
|
|
MTT reagent
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
NSAIDs
|
non-steroidal anti-inflammatory
drugs
|
References
|
1
|
Ferlay J, Parkin DM and Steliarova-Foucher
E: Estimates of cancer incidence and mortality in Europe in 2008.
Eur J Cancer. 46:765–781. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thun MJ, Namboodiri MM and Heath CW Jr:
Aspirin use and reduced risk of fatal colon cancer. N Engl J Med.
325:1593–1596. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Logan RF, Little J, Hawtin PG and
Hardcastle JD: Effect of aspirin and non-steroidal
anti-inflammatory drugs on colorectal adenomas: case-control study
of subjects participating in the Nottingham faecal occult blood
screening programme. BMJ. 307:285–289. 1993. View Article : Google Scholar
|
|
4
|
Baron JA, Cole BF, Sandler RS, et al: A
randomized trial of aspirin to prevent colorectal adenomas. N Engl
J Med. 348:891–899. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Din FV, Theodoratou E, Farrington SM, et
al: Effect of aspirin and NSAIDs on risk and survival from
colorectal cancer. Gut. 59:1670–1679. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
La Vecchia C, Negri E, Franceschi S, et
al: Aspirin and colorectal cancer. Br J Cancer. 76:675–677.
1997.
|
|
7
|
Corpet DE and Pierre F: Point: from animal
models to prevention of colon cancer. Systematic review of
chemoprevention in min mice and choice of the model system. Cancer
Epidemiol Biomarkers Prev. 12:391–400. 2003.PubMed/NCBI
|
|
8
|
Liu Y, Ju J, Xiao H, et al: Effects of
combination of calcium and aspirin on azoxymethane-induced aberrant
crypt foci formation in the colons of mice and rats. Nutr Cancer.
60:660–665. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bousserouel S, Gosse F, Bouhadjar M, Soler
L, Marescaux J and Raul F: Long-term administration of aspirin
inhibits tumour formation and triggers anti-neoplastic molecular
changes in a pre-clinical model of colon carcinogenesis. Oncol Rep.
23:511–517. 2010.
|
|
10
|
Sansom OJ, Stark LA, Dunlop MG and Clarke
AR: Suppression of intestinal and mammary neoplasia by lifetime
administration of aspirin in Apc(Min/+) and Apc(Min/+), Msh2(−/−)
mice. Cancer Res. 61:7060–7064. 2001.PubMed/NCBI
|
|
11
|
Lal G, Ash C, Hay K, et al: Suppression of
intestinal polyps in Msh2-deficient and non-Msh2-deficient multiple
intestinal neoplasia mice by a specific cyclooxygenase-2 inhibitor
and by a dual cyclooxygenase-1/2 inhibitor. Cancer Res.
61:6131–6136. 2001.PubMed/NCBI
|
|
12
|
Rothwell PM, Fowkes FG, Belch JF, Ogawa H,
Warlow CP and Meade TW: Effect of daily aspirin on long-term risk
of death due to cancer: analysis of individual patient data from
randomised trials. Lancet. 377:31–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Burn J, Gerdes AM, Macrae F, et al:
Long-term effect of aspirin on cancer risk in carriers of
hereditary colorectal cancer: an analysis from the CAPP2 randomised
controlled trial. Lancet. 378:2081–2087. 2011. View Article : Google Scholar
|
|
14
|
Rothwell PM, Wilson M, Price JF, Belch JF,
Meade TW and Mehta Z: Effect of daily aspirin on risk of cancer
metastasis: a study of incident cancers during randomised
controlled trials. Lancet. 379:1591–1601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chan AT, Ogino S and Fuchs CS: Aspirin use
and survival after diagnosis of colorectal cancer. JAMA.
302:649–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laine L: The gastrointestinal effects of
nonselective NSAIDs and COX-2-selective inhibitors. Semin Arthritis
Rheum. 32:25–32. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gasche C, Goel A, Natarajan L and Boland
CR: Mesalazine improves replication fidelity in cultured colorectal
cells. Cancer Res. 65:3993–3997. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Campregher C, Honeder C, Chung H,
Carethers JM and Gasche C: Mesalazine reduces mutations in
transforming growth factor β receptor II and activin type II
receptor by improvement of replication fidelity in mononucleotide
repeats. Clin Cancer Res. 16:1950–1956. 2010.PubMed/NCBI
|
|
19
|
McIlhatton MA, Tyler J, Burkholder S, et
al: Nitric oxide-donating aspirin derivatives suppress
microsatellite instability in mismatch repair-deficient and
hereditary nonpolyposis colorectal cancer cells. Cancer Res.
67:10966–10975. 2007. View Article : Google Scholar
|
|
20
|
Tesei A, Zoli W, Fabbri F, et al: NCX
4040, an NO-donating acetylsalicylic acid derivative: efficacy and
mechanisms of action in cancer cells. Nitric Oxide. 19:225–236.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Selvendiran K, Bratasz A, Tong L, Ignarro
LJ and Kuppusamy P: NCX-4016, a nitro-derivative of aspirin,
inhibits EGFR and STAT3 signaling and modulates Bcl-2 proteins in
cisplatin-resistant human ovarian cancer cells and xenografts. Cell
Cycle. 7:81–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao W, Mackenzie GG, Murray OT, Zhang Z
and Rigas B: Phosphoaspirin (MDC-43), a novel benzyl ester of
aspirin, inhibits the growth of human cancer cell lines more
potently than aspirin: a redox-dependent effect. Carcinogenesis.
30:512–519. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang D and DuBois RN: Pro-inflammatory
prostaglandins and progression of colorectal cancer. Cancer Lett.
267:197–203. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sheng H, Shao J, Morrow JD, Beauchamp RD
and DuBois RN: Modulation of apoptosis and Bcl-2 expression by
prostaglandin E2 in human colon cancer cells. Cancer Res.
58:362–366. 1998.PubMed/NCBI
|
|
25
|
Harris RE, Beebe-Donk J and Alshafie GA:
Similar reductions in the risk of human colon cancer by selective
and nonselective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer.
8:2372008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Elwood PC, Gallagher AM, Duthie GG, Mur LA
and Morgan G: Aspirin, salicylates, and cancer. Lancet.
373:1301–1309. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dibra HK, Perry CJ and Nicholl ID:
Non-steroidal anti-inflammatory drugs, DNA repair and cancer. DNA
Repair and Human Health. Vengrova S: InTech; pp. 743–776. 2011,
http://cdn.intechopen.com/pdfs-wm/22182.pdf.
|
|
28
|
Kopp E and Ghosh S: Inhibition of NF-kappa
B by sodium salicylate and aspirin. Science. 265:956–959. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thoms HC, Dunlop MG and Stark LA:
p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4
stimulates nucleolar translocation of RelA and apoptosis in
colorectal cancer cells. Cancer Res. 67:1660–1669. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu HG, Huang JA, Yang YN, et al: The
effects of acetylsalicylic acid on proliferation, apoptosis, and
invasion of cyclooxygenase-2 negative colon cancer cells. Eur J
Clin Invest. 32:838–846. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luciani MG, Campregher C and Gasche C:
Aspirin blocks proliferation in colon cells by inducing a G1 arrest
and apoptosis through activation of the checkpoint kinase ATM.
Carcinogenesis. 28:2207–2217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shureiqi I, Chen D, Lotan R, et al:
15-Lipoxygenase-1 mediates nonsteroidal anti-inflammatory
drug-induced apoptosis independently of cyclooxygenase-2 in colon
cancer cells. Cancer Res. 60:6846–6850. 2000.PubMed/NCBI
|
|
33
|
Goel A, Chang DK, Ricciardiello L, Gasche
C and Boland CR: A novel mechanism for aspirin-mediated growth
inhibition of human colon cancer cells. Clin Cancer Res. 9:383–390.
2003.PubMed/NCBI
|
|
34
|
Pangburn HA, Kraus H, Ahnen DJ and Rice
PL: Sulindac metabolites inhibit epidermal growth factor receptor
activation and expression. J Carcinog. 4:162005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pangburn HA, Ahnen DJ and Rice PL:
Sulindac metabolites induce proteosomal and lysosomal degradation
of the epidermal growth factor receptor. Cancer Prev Res.
3:560–572. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ruschoff J, Wallinger S, Dietmaier W, et
al: Aspirin suppresses the mutator phenotype associated with
hereditary nonpolyposis colorectal cancer by genetic selection.
Proc Natl Acad Sci USA. 95:11301–11306. 1998. View Article : Google Scholar
|
|
37
|
Deb J, Dibra H, Shan S, et al: Activity of
aspirin analogues and vanillin in a human colorectal cancer cell
line. Oncol Rep. 26:557–565. 2011.PubMed/NCBI
|
|
38
|
Hussey HJ and Tisdale MJ: Effect of
polyunsaturated fatty acids on the growth of murine colon
adenocarcinomas in vitro and in vivo. Br J Cancer. 70:6–10. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Carmichael J, DeGraff WG, Gazdar AF, Minna
JD and Mitchell JB: Evaluation of a tetrazolium-based semiautomated
colorimetric assay: assessment of chemosensitivity testing. Cancer
Res. 47:936–942. 1987.PubMed/NCBI
|
|
41
|
Stark LA and Dunlop MG: Nucleolar
sequestration of RelA (p65) regulates NF-κB-driven transcription
and apoptosis. Mol Cell Biol. 25:5985–6004. 2005.PubMed/NCBI
|
|
42
|
Stark LA, Din FV, Zwacka RM and Dunlop MG:
Aspirin-induced activation of the NF-κB signaling pathway: a novel
mechanism for aspirin-mediated apoptosis in colon cancer cells.
FASEB J. 15:1273–1275. 2001.
|
|
43
|
Loveridge CJ, MacDonald AD, Thoms HC,
Dunlop MG and Stark LA: The proapoptotic effects of sulindac,
sulindac sulfone and indomethacin are mediated by nucleolar
translocation of the RelA(p65) subunit of NF-κB. Oncogene.
27:2648–2655. 2008.PubMed/NCBI
|
|
44
|
Campbell KJ, Rocha S and Perkins ND:
Active repression of antiapoptotic gene expression by RelA(p65)
NF-κB. Mol Cell. 13:853–865. 2004.PubMed/NCBI
|
|
45
|
Walder JA, Zaugg RH, Walder RY, Steele JM
and Klotz IM: Diaspirins that cross-link beta chains of hemoglobin:
bis(3,5-dibromosalicyl) succinate and bis(3,5-dibromosalicyl)
fumarate. Biochemistry. 18:4265–4270. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Alfonso LF, Srivenugopal KS and Bhat GJ:
Does aspirin acetylate multiple cellular proteins? (Review). Mol
Med Rep. 2:533–537. 2009.PubMed/NCBI
|
|
47
|
Schwenger P, Alpert D, Skolnik EY and
Vilcek J: Cell typespecific activation of c-Jun N-terminal kinase
by salicylates. J Cell Physiol. 179:109–114. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kojima M, Morisaki T, Sasaki N, et al:
Increased nuclear factor-kB activation in human colorectal
carcinoma and its correlation with tumor progression. Anticancer
Res. 24:675–681. 2004.PubMed/NCBI
|
|
49
|
Greenspan EJ, Madigan JP, Boardman LA and
Rosenberg DW: Ibuprofen inhibits activation of nuclear β-catenin in
human colon adenomas and induces the phosphorylation of GSK-3β.
Cancer Prev Res. 4:161–171. 2011.
|
|
50
|
Thoms HC, Loveridge CJ, Simpson J, et al:
Nucleolar targeting of RelA(p65) is regulated by COMMD1-dependent
ubiquitination. Cancer Res. 70:139–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Meltzer PC, Blundell P, Yong YF, et al:
2-Carbomethoxy-3-aryl-8-bicyclo[3.2.1]octanes: potent non-nitrogen
inhibitors of monoamine transporters. J Med Chem. 43:2982–2991.
2000.PubMed/NCBI
|



















