Introduction
In developed countries, bladder cancer is one of the
most common forms of tumor that often lead to mortality in men
(1). The proliferation and
metastasis of bladder cancer cells plays an essential role in the
development and progression of bladder tumors (2). The proliferation of bladder cancer
cells has been definitively associated with the induction of
multiple mitogen-activated protein kinases (MAPKs), including
extracellular signal regulated kinase (ERK1/2), c-Jun NH2-terminal
kinase (JNK), and p38 MAPK (p38) (3). In addition, it is widely accepted that
the progression of the cell cycle is critical for the development
and progression of bladder tumor cells (4,5).
Tumor metastasis has been implicated in the
migration and invasion of bladder cancer cells (2,6,7). The
expression of matrix metalloproteinase-9 (MMP-9) is known to be
involved in the migration and invasion of bladder tumor cells
(2,6,7).
Previous reports, based on in vitro and in vivo
studies, have shown that MMP-9 is critical for the progression of
bladder tumors via its regulation of both migration and invasion by
bladder cancer cells (2,6,7). Many
studies have shown that MMP-9 expression is regulated primarily by
transcription factors NF-κB, AP-1, and Sp-1 (7–10).
Recent studies have shown that MAPKs are connected to the
expression of MMP-9 in bladder cancer cells via the activation of
NF-κB and AP-1 (7,11).
Human erythropoietin (EPO) plays a role in the
control of red blood cell production (12,13).
EPO stimulates the activation of JAK2/STAT5, MAPKs,
phosphatidylinositol 3-kinase (PI3K), and Akt signaling through the
binding of human erythropoietin receptor (EPOR) (14). Several studies have shown that the
exogenous treatment of EPO and the overexpression of EPOR stimulate
proliferation, migration and invasiveness in various cell types
(15–19). On the other hand, other reports have
not associated EPO with a proliferative effect on cancer cells
(20–22). Although many studies have
investigated the role of EPO in cancer cells, the precise molecular
mechanisms underlying the cell-cycle regulation and signaling
pathways that are linked with cell proliferation, migration and
invasion of cancer cells remain unclear.
Recently, we found an upregulated gene expression
level of EPO in bladder tumor patients using microarray expression
analysis (23). The purpose of the
present study was to determine the role and regulatory mechanisms
of the EPO gene in bladder cancer cells. In this study, we
identified a novel mechanism of cell-cycle inhibitor p21WAF1 for
the induction of proliferation, migration and invasion in EPO gene
expression in bladder cancer cells.
Materials and methods
Materials
Polyclonal antibodies to ERK, phospho-ERK, p38MAPK,
phospho-p38MAPK, JNK, and phospho-JNK were obtained from Cell
Signaling (Danvers, MA, USA). Polyclonal antibodies to cyclin E,
CDK2, CDK4, cyclin D1, p53, p21WAF1, p27KIP1 and GAPDH were
purchased from Santa Cruz (Santa Cruz, CA, USA). U0126 was obtained
from Calbiochem (San Diego, CA, USA). Polyclonal MMP-9 antibody was
obtained from Chemicon (Temecula, CA, USA). Small interfering RNA
(siRNA) oligonucleotides targeting p21WAF1 and scramble were
designed and synthesized by Dharmacon (Lafayette, CO, USA).
pcDNA3-EPO plasmid containing human wild-type full-length EPO was
generously provided by Dr Taiho Kambe (Kyoto University,
Japan).
Cell cultures
A human bladder carcinoma cell line (5637) was
obtained from the American Type Culture Collection. The cells were
maintained in DMEM (4.5 g glucose/liter) and were supplemented with
10% fetal calf serum, L-glutamine, and antibiotics (Biological
Industries, Beit Haemek, Israel) at 37°C in a 5% CO2
humidified incubator.
Cell transfection
5637 cells were transfected with pcDNA3-EPO (EPO) or
pcDNA3 (no insert, EV) in 100-mm dishes using the Superfect reagent
(Qiagen, Valencia, CA, USA) according to the manufacturer’s
protocol. After 24 h, cells were split at a 1:5 dilution and
exposed for 2–3 weeks in G418 (Boehringer Mannheim, Indianapolis,
IN, USA)-containing medium (800 μg/ml), and the resultant colonies
were selected based on their resistance to G418. The expression of
EPO was confirmed by immunoblot analysis using a monoclonal
antibody against EPO (R&D Systems).
[3H]thymidine
incorporation
For [3H]thymidine-uptake experiments,
cells were grown to near confluence in 24-well tissue culture
plates and were then labeled with [methyl-3H]thymidine
(New England Nuclear, Boston, MA, USA) at 1 μCi/ml. After labeling,
the cells were cultured for 24 h and washed with phosphate-buffered
saline, fixed in cold 10% trichloroacetic acid, then washed with
95% ethanol. Incorporated [3H]thymidine was extracted in
0.2 M NaOH and measured using a liquid scintillation counter as
previously described (24).
Immunoblotting
The cells were incubated with 10% FBS for various
durations at 37°C. The cells were then washed twice with cold PBS
and freeze-thawed in 250 μl lysis buffer [containing, in mmol/l,
HEPES (pH 7.5) 50, NaCl 150, EDTA 1, EGTA 2.5, DTT 1,
β-glycerophosphate 10, NaF 1, Na3VO4 0.1, and
phenylmethylsulfonyl fluoride 0.1 and 10% glycerol, 0.1% Tween-20,
10 μg/ml of leupeptin, and 2 μg/ml of aprotinin], and then scraped
into 1.5-ml tubes. The lysates were placed on ice for 15 min and
then centrifuged at 12,000 rpm for 20 min at 4°C. The protein
concentration of the supernatant was determined using the Bradford
reagent method (Bio-Rad). Equal amounts of cellular proteins were
resolved by electrophoresis on a 0.1% SDS-10% polyacrylamide gel
(SDS-PAGE) under denaturing conditions. The proteins were
transferred electrophoretically to nitrocellulose membranes
(Hybond; Amersham Corp). After blocking in 10 mmol/l Tris-HCl (pH
8.0), 150 mmol/l NaCl, and 5% (wt/vol) nonfat dry milk, the
membranes were treated with primary antibodies for 90 min, followed
by incubation with peroxidase-conjugated secondary antibodies for
45 min. The immune complexes were detected using a
chemiluminescence reagent kit (Amersham Corp.). For the
immunoblotting studies, the experiments were repeated at least 3
times.
Immunoprecipitation and immune complex
kinase assays
Cell lysates were prepared with ice-cold lysis
buffer [containing, in mM/l, HEPES (pH 7.5) 50, NaCl 150, EDTA 1,
EGTA 2.5, DTT 1, β-glycerophosphate 10, NaF 1,
Na3VO4 0.1, and phenylmethylsulfonyl fluoride
0.1 and 10% glycerol, 0.1% Tween-20, 10 μg/ml of leupeptin, and 2
μg/ml of aprotinin] and sonicated at 4°C [Micro ultrasonic cell
disruptor (Kontes), 30% power, twice for 10 sec each time]. Lysates
were clarified by centrifugation at 10,000 × g for 5 min, and the
supernatants were precipitated by treatment with protein
A-Sepharose beads precoated with saturating amounts of the
indicated antibodies at 4°C for 2 h. When monoclonal antibodies
were used, protein A-Sepharose was pretreated with rabbit
anti-mouse immunoglobulin G (Jackson Immuno Research Laboratories).
The immunoprecipitated proteins on the beads were washed 4 times
with 1 ml of lysis buffer and twice with a kinase buffer
(containing, in mM/l, HEPES 50, MgCl2 10, DTT 1,
β-glycerophosphate 10, NaF 1, and sodium orthovanadate 0.1). The
final pellet was resuspended in 25 μl of kinase buffer containing
either 1 μg of glutathione S-transferase (GST)-pRb C-terminal (pRb
amino acids 769 to 921) fusion protein (Santa Cruz Biotechnology)
or 5 μg of histone H1 (Life Technologies, Inc.), 20 μM/l ATP, and 5
μCi of [γ32P]ATP (4,500 μCi/mmol; ICN), and were then
incubated for 20 min at 30°C with occasional mixing. The reaction
was terminated by the addition of 25 μl of 2X concentrated Laemmli
sample buffer and separated on either 10 or 12.5%
SDS-polyacrylamide gels. The migration of histone H1 or GST-pRb was
determined by Coomassie blue staining. Phosphorylated pRb and
histone H1 were visualized.
Wound-healing migration assay
Cells were plated on 6-well dishes and grown to 90%
confluency in 2 ml of growth medium. The cells were damaged using a
2-mm-wide tip. They were allowed to migrate, and images were
captured through an inverted microscope (magnification, ×40).
Invasion assay
Cells (2.5×104) were resuspended with 100
μl of medium and placed in the upper part of a Transwell plate. The
cells were then incubated for 24 h. The cells had to pass through a
polycarbonate membrane with 8-μm-sized pores and a thin layer of an
ECM Matrix-like material. The ability of the cells to invade the
ECM Matrix-like material was determined using a commercial cell
invasion assay kit (Chemicon International, Billerica, MA,
USA).
Zymography
Conditioned medium was electrophoresed in a
polyacrylamide gel containing 1 mg/ml gelatin. The gel was then
washed at room temperature for 2 h with 2.5% Triton X-100 and
subsequently incubated at 37°C overnight in a buffer containing 10
mM CaCl2, 150 mM NaCl, and 50 mM Tris-HCl, pH 7.5. The
gel was stained with 0.2% Coomassie blue and photographed on a
light box. Proteolysis was detected as a white zone in a dark blue
field (7,9,24).
Nuclear extracts and electrophoretic
mobility shift assay (EMSA)
Cultured cells were collected by centrifugation,
washed and suspended in a buffer containing 10 mM Hepes (pH 7.9),
10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, and 0.5 mM PMSF.
After 15 min on ice, the cells were vortexed in the presence of
0.5% Nonidet NP-40. The nuclear pellet was then collected by
centrifugation and extracted in a buffer containing 20 mM Hepes pH
7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 1 mM PMSF for
15 min at 4°C.
The nuclear extract (10–20 μg) was preincubated at
4°C for 30 min with a 100-fold excess of an unlabeled
oligonucleotide spanning the -79 MMP-9 cis element of
interest. The sequences were: AP-1, CTGACCCCTGAGTCAGCACTT; NF-κB,
CAGTGGAATTCCCCAGCC; and Sp-1, GCCCATT CCTTCCGCCCCCAGATGAAGCAG. The
reaction mixture was then incubated at 4°C for 20 min in a buffer
(25 mM HEPES buffer pH 7.9, 0.5 mM EDTA, 0.5 mM DTT, 0.05 M NaCl,
and 2.5% glycerol) with 2 μg of poly dI/dC and 5 fmol
(2×104 cpm) of a Klenow end-labeled (32P-ATP)
30-mer oligonucleotide, which spanned the DNA binding site in the
MMP-9 promoter. The reaction mixture was electrophoresed at 4°C in
a 6% polyacrylamide gel using a TBE (89 mM Tris, 89 mM boric acid
and 1 mM EDTA) running buffer. The gel was rinsed with water, dried
and exposed to X-ray film overnight (7,9,24).
Statistical analysis
Where appropriate, data are expressed as the mean ±
SE. Data were analyzed by factorial ANOVA and a Fisher’s least
significant difference test where appropriate. Statistical
significance was set at P<0.05.
Results
EPO gene overexpression induces DNA
synthesis, migration and invasion of bladder cancer 5637 cells
To investigate the role of EPO in the proliferation,
migration and invasion of cancer cells, bladder cancer 5637 cells
were transfected with an EPO cDNA (EPO) or an identical empty
vector lacking a cDNA insert as a control (EV). Stable cell clones
(EPO) were isolated, and parental cells (UN) were used as an
additional control. Validation of the EPO gene was evaluated by
immunoblotting (Fig. 1A). Cells
expressing EPO showed an ~2.1-fold increase in [3H]
thymidine incorporation, compared to UN and EV transfectants
(Fig. 1B). We then investigated the
effect of the EPO gene on the migratory and invasive capacity of
bladder cancer 5637 cells using a wound-healing migration and
invasion assay system. Compared with UN and EV transfectants, EPO
gene transfectants significantly increased the migratory and
invasive potential of cells (Fig. 1C
and D).
EPO gene transfectants stimulate MMP-9
expression via the activation of transcription factor NF-κB, Sp-1
and AP-1
Gelatinolytic zymography assay was employed to study
the systemic mechanism of the migration and invasion of 5637 cells
expressing EPO. Compared with that of the UN, the conditioned media
from the EPO gene transfectant cells induced the upregulation of
MMP-9 gelatinolytic activity (Fig.
1E). In addition, EV transfectants had no effect on MMP-9
activity (Fig. 1E). Similar results
were detected in immunoblot analysis (Fig. 1E). To determine whether the
stimulatory effect of the EPO gene on MMP-9 expression was mediated
through three types of motifs, NF-κB, Sp-1, and AP-1 activation,
EMSA was next performed with the nuclear extracts of UN, EV
transfectant and EPO gene transfectant cells. Nuclear extracts from
EPO gene transfectants showed increased binding activities of the
NF-κB, Sp-1 and AP-1 motifs (Fig.
1F). However, neither the EV transfectants nor that of the UN
had any effect on NF-κB, Sp-1 or AP-1 binding activities (Fig. 1F).
EPO gene transfectants stimulate the
phosphorylation of ERK1/2 in 5637 cells
To assess the effect of the signaling pathways
involved in the proliferation of EPO gene transfectants, ERK1/2,
p38MAPK and JNK phosphorylations were analyzed. As shown in
Fig. 2A, the overexpression of the
EPO gene induced an increased phosphorylation of ERK1/2. In
addition, pre-treatment with U0126 (ERK1/2-specific inhibitor)
suppressed ERK1/2 phosphorylation in EPO gene transfectant cells
(Fig. 2B). However, EPO gene
transfectants did not stimulate p38MAPK and JNK phosphorylation
(Fig. 2A).
U0126 inhibits the potential for
proliferation, migration, and invasion in EPO gene
transfectants
Since the overexpression of EPO stimulated ERK1/2
phosphorylation, we investigated the role of ERK1/2 in the
EPO-stimulated proliferation, migration and invasion of 5637 cells.
The increased cell proliferation after EPO gene transfection was
strongly reversed in the presence of U0126 (Fig. 2C). In addition, treatment with U0126
blocked the EPO-induced wound healing migration and invasion of
5637 cells (Fig. 2D and E).
Moreover, the U0126 treatment reversed MMP-9 gelatinolytic activity
in the conditioned media from EPO gene transfectant cells to the
level of EV transfectants (Fig.
2F). Similar results were observed in immunoblot analysis
(Fig. 2F). To further verify the
regulatory mechanism of MMP-9 expression, we next performed an EMSA
experiment. As shown in Fig. 2G,
the increased NF-κB binding activation in EPO gene transfectants
was reversed to basal levels of EV transfectants in the presence of
U0126. By contrast, U0126 treatment had no significant effect on
the binding activity of Sp-1 and AP-1 in EPO gene transfectants
(Fig. 2G).
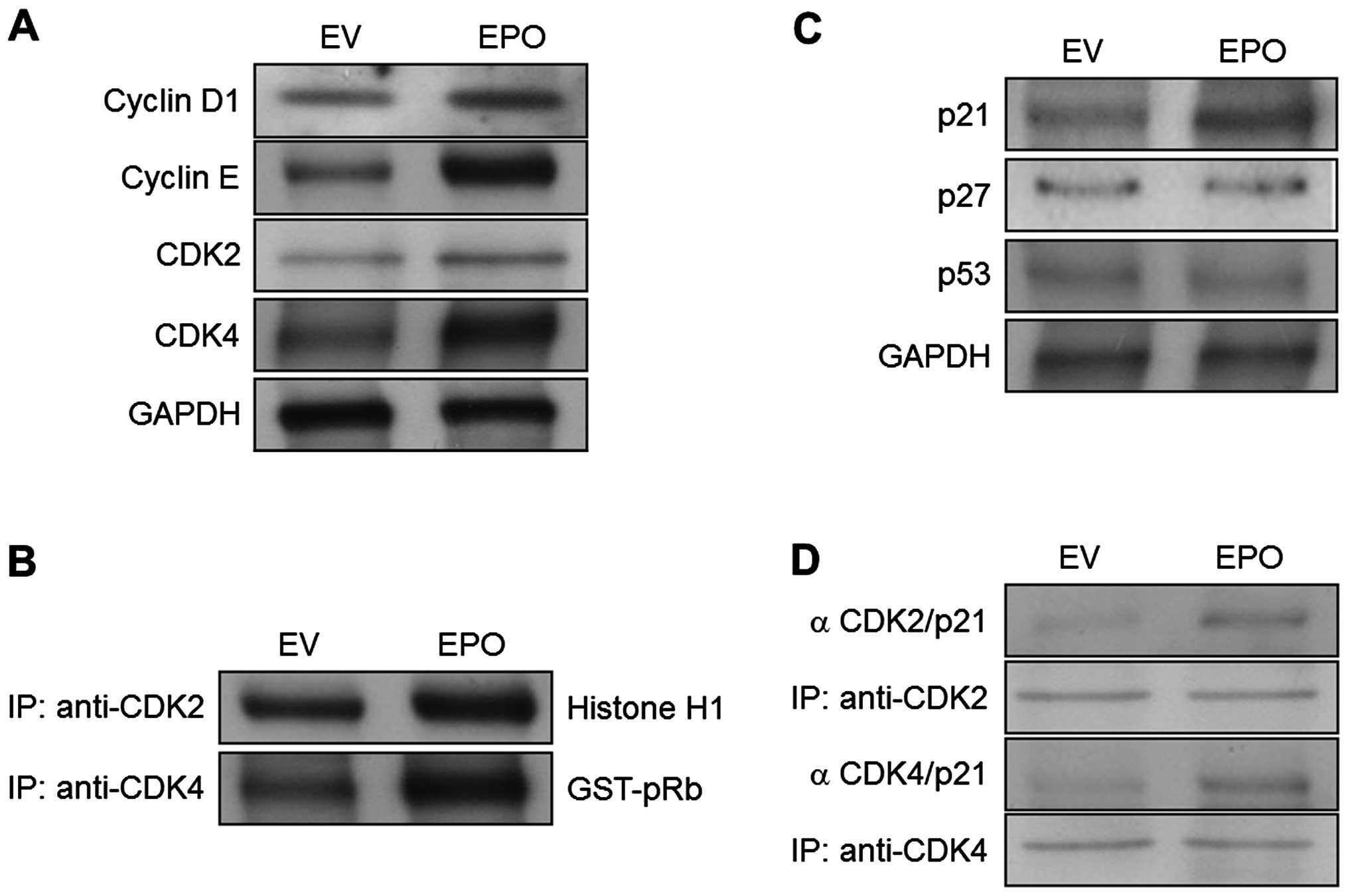
EPO gene transfectants modulate G1
cell-cycle-associated proteins and induce p21WAF1 expression
To verify that the observed growth stimulatory
effects of EPO gene transfectants were involved in the increased
expression of cell-cycle machinery, the levels of cell-cycle
machinery molecules were investigated. The expression levels of the
G1-associated factors, cyclin D1/CDK4 and cyclin E/CDK2, were
upregulated in EPO gene transfectants by comparison with the EV
transfectants (Fig. 3A). In
addition, overexpression of the EPO gene resulted in a significant
increase in the kinase activities of both CDK2- and
CDK4-immunoprecipitates compared with the EV transfectant cells
(Fig. 3B). Since the cell-cycle
inhibitors are known to regulate the G1- to S-phase transition
checkpoints, the expression levels of p21WAF1 and p27KIP1 were
assessed in both EPO gene transfectants and EV transfectants.
Notably, the results showed that p21WAF1 levels were increased in
EPO gene-transfected cells compared with EV-transfected cells
(Fig. 3C). However, under similar
experimental conditions, the expression of p27KIP1 was unaffected
(Fig. 3C). Moreover, the
overexpression of the EPO gene had no effect on the induction of
the tumor suppressor protein p53 (Fig.
3C). We next examined the effects of the EPO gene on
interactions between p21WAF1 and CDKs. Immunoprecipitation analysis
revealed that the EPO gene transfectants exhibited a strong
increase in the association of p21WAF1 and CDK2 compared with the
EV transfectants (Fig. 3D). The
binding of p21WAF1 and CDK4 was also increased in the EPO gene
transfectants (Fig. 3D).
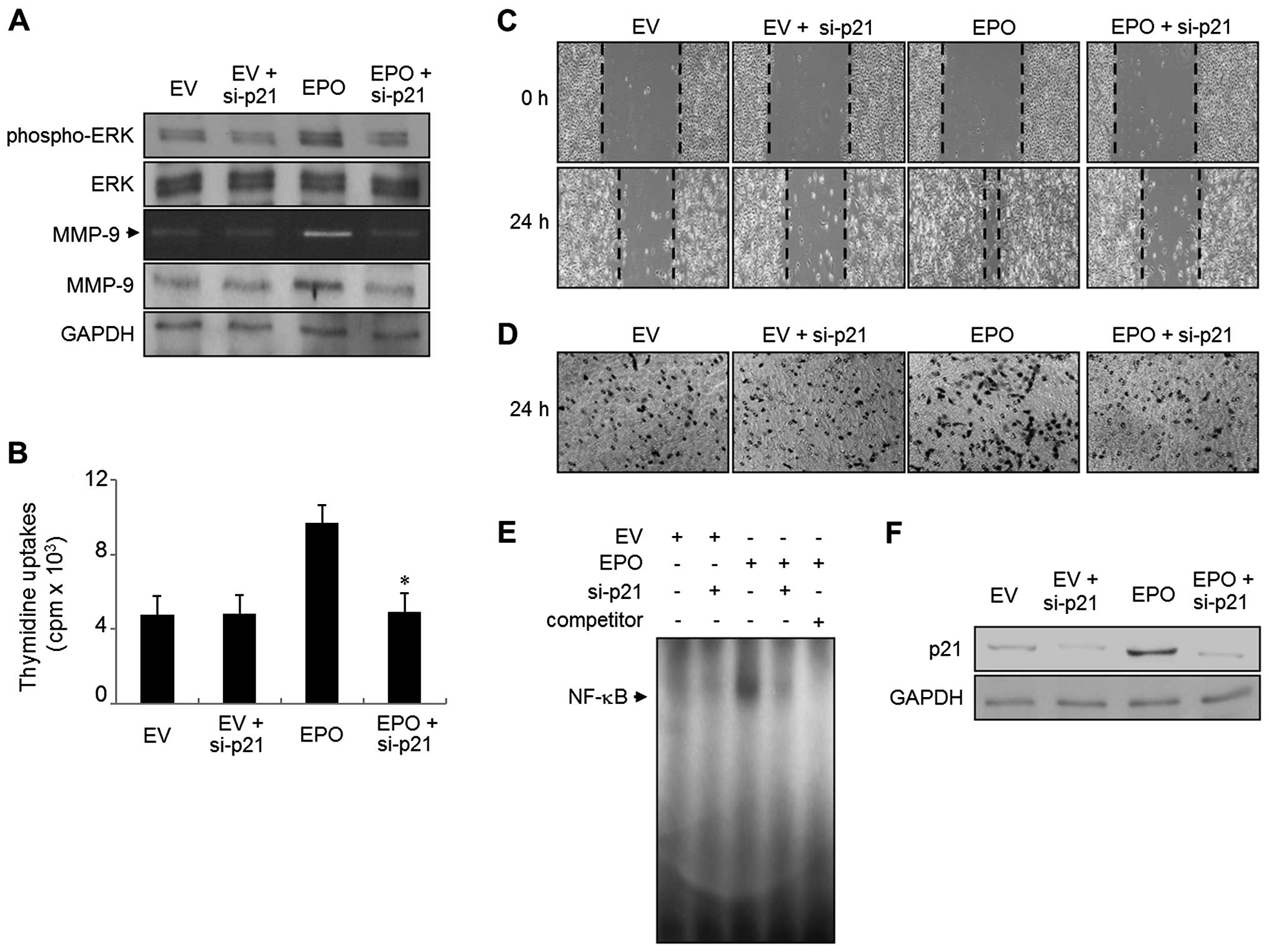
siRNA-mediated knockdown of p21WAF1
restores the proliferation, migration, invasion, MMP-9 expression,
ERK1/2 phosphorylation and binding activity of NF-κB in the EPO
gene transfectants
To determine the effect of inhibiting the p21WAF1
expression in EPO gene transfectants, we used either a
p21WAF1-specific siRNA (si-p21) or a scrambled siRNA. The
expression level of p21WAF1 was determined by immunoblot analysis
(Figs. 4F and 5F). As shown in Fig. 4A and B, EPO gene transfectants of
si-p21 suppressed the increase in proliferation and ERK1/2
phosphorylation. EPO-induced wound-healing migration and invasive
ability was also reversed in the presence of si-p21 transfection
(Fig. 4C and D). In addition, the
upregulation of MMP-9 expression was abolished in EPO cells
transfected with si-p21, as evidenced by gelatin zymography and
immunoblot analysis (Fig. 4A).
Finally, NF-κB DNA binding activity was almost impeded by the
transfection of si-p21 in EPO gene transfectants (Fig. 4E). EPO gene transfectants had no
significant effect on the proliferation, migration, invasion, MMP-9
expression, ERK1/2 phosphorylation and binding activity of NF-κB in
the presence of scrambled siRNA transfection (Fig. 5A–E).
Discussion
In a previous study, our gene expression profile
data produced by cDNA microarray experimentation showed that the
expression levels of the EPO gene were increased in patients with
bladder tumors (23). In the
present study, we suggest a novel role for the EPO gene that is
involved in the p21WAF1-mediated proliferation, migration and
invasion of bladder cancer 5637 cells.
In the past few years, a number of studies on the
modulation of cell proliferation and migration by EPO have been
conducted (15–19). Published evidence has indicated that
EPO may be involved in the differentiation of erythroid progenitor
cells in vitro (12–14). However, previous studies have also
shown that EPO did not stimulate the growth of tumor cell lines
(20–22). In the present study, transfection of
the EPO gene into bladder cancer 5637 cells resulted in increased
thymidine uptake at the basal level. In addition, overexpression of
the EPO gene induced the migration and invasion of 5637 cells.
Consistent with our present results, several studies have reported
that exogenous EPO or EPO gene expression induced the
proliferation, migration and invasion of cancer cell lines
(15–19). These results suggest that
overexpression of the EPO gene is involved in the induction of the
proliferation, migration and invasion of bladder cancer 5637
cells.
The elevated level of MMP-9 expression was strongly
associated with the progression and migration of bladder cancer in
animal and clinical studies (2,6,7). In
results from the present study, overexpression of the EPO gene
stimulated an enhanced level of MMP-9 expression. Moreover, we
analyzed the transcriptional regulation of MMP-9 in 5637 cells
expressing the EPO gene, and found that it promoted MMP-9
expression via the increased binding activation of NF-κB, Sp-1 and
AP-1 in 5637 cells. These results suggest that the EPO induced the
expression of MMP-9 via the activation of NF-κB, Sp-1 and AP-1 in
5637 cells, which caused the destruction of ECM and influenced the
migration and invasion of bladder cancer progression.
Since transfection of the EPO gene into 5637 cells
resulted in increased thymidine uptake, we next investigated the
MAPK signaling pathways, including ERK1/2, JNK, and p38 MAPK, in
EPO gene transfectants. Many studies have suggested the stimulatory
effects of EPO on ERK1/2 phosphorylation in several lines of cells
(14,18). Consistent with the results of
previous studies, in the present study EPO gene expression induced
ERK1/2 phosphorylation without altering the phosphorylation of
either JNK or p38MAPK. In addition, an inhibitor of the ERK1/2
signaling molecule U0126 inhibited the proliferation, migration and
invasion of cells transfected by the EPO gene. Moreover, U0126
suppressed both the expression of MMP-9 and the activation of NF-κB
in EPO gene transfectants. Our results demonstrated that ERK1/2
signaling is involved in the proliferation, migration and invasion
of EPO gene-expressed 5637 cells. We also found that transcription
factor NF-κB is important in the ERK1/2-mediated regulation of
MMP-9 expression in 5637 bladder cancer cells expressing the EPO
gene.
The G1- to S-phase cell-cycle progression is a key
event in the progression and development of tumor cells (25,26).
The G1- to S-phase transition is regulated by cyclin-dependent
kinases(CDKs), including cyclin D1/CDK4 and cyclin E/CDK2, which
are negatively controlled via the binding of CDK inhibitors (CKIs)
such as p21WAF1 and p27KIP1 (25,26).
Several studies have suggested the involvement of EPO in cell-cycle
regulators during the differentiation of erythroid progenitor cells
and the proliferation of hematopoietic cells (27,28).
However, the molecular regulation of cell-cycle regulators
coordinated with the proliferation, migration and invasion of tumor
cells merits further investigation. Therefore, we examined the
effect of the EPO gene on CDK and CKI levels responsible for the G1
to S transition. Results of the present study showed that
overexpression of the EPO gene in 5637 cells significantly
upregulated cyclin D1, cyclin E, CDK2 and CDK4, along with the
stimulation of CDK4 and CDK2 kinase activity. Notably, our data
also showed a significantly elevated level of p21WAF1, but not of
p27KIP1 and p53. To further elucidate how p21WAF1 modulates cell
proliferation, migration and invasion in EPO gene transfectants, we
conducted a specific siRNA knockdown experiment using p21WAF1
(si-p21). Our data suggest the novel theory that p21WAF1 is
indispensable for the proliferation, migration and invasion of 5637
cells in response to the EPO gene. Our results further enhanced
these findings by demonstrating that p21WAF1 regulated
ERK/1/2-coordinated MMP-9 expression via upregulated NF-κB binding
in the EPO gene-induced proliferation, migration and invasion of
bladder cancer 5637 cells.
Many studies have demonstrated that p21WAF1 is a
negative regulator in the control of cell-cycle progression
(25,26), but previous studies have suggested
that it is a positive modulator in the proliferation and migration
of several cell lines (7,23,25,26).
Several studies now associate p21WAF1 with clinical evidence that
shows involvement in bladder cancer stage, progression and
prognosis (29,30). Although the significant role of
cell-cycle regulation has been reported in the progression and
development of tumors (29–31), the molecular mechanism of p21WAF1
regarding EPO expression in tumor progression remains to be fully
explored. Results from the present study propose the novel
conclusion that p21WAF1 may provide an important role in the
proliferation, migration and invasion of bladder cancer cells that
is induced by the EPO gene.
Taken together, the evidence suggests that the EPO
gene induces the proliferation, G1- to S-phase cell-cycle
progression, migration and invasion of bladder cancer 5637 cells.
In addition, the results of the present study demonstrate the novel
concept that the well-known cell-cycle inhibitor p21WAF1 is
required for cell proliferation, migration and invasion through
ERK1/2-mediated MMP-9 expression by stimulating NF-κB binding
activity in bladder cancer cells that express the EPO gene. In
conclusion, results of the present study indicate that bladder
cancer cells induced by the EPO gene are associated with
proliferation, migration and invasion that contribute to the
progression of bladder tumor, making it an effective target
candidate of potential therapies for the prevention and treatment
of malignant cells.
Acknowledgements
This research was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF), funded by the Ministry of Education, Science and Technology
(2008–0062611).
References
|
1
|
Jemal A, Siegel R, Ward E, Murray E, Xu T
and Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 57:43–66.
2007. View Article : Google Scholar
|
|
2
|
Black PC and Dinney CP: Bladder cancer
angiogenesis and metastasis - translation from murine model to
clinical trial. Cancer Metastasis Rev. 26:623–634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zachos I, Konstantinopoulos PA, Tzortzis
V, Gravas S, Karatzas A, Karamouzis MV, Melekos M and Papavassiliou
AG: Systemic therapy of metastatic bladder cancer in the molecular
era: current status and future promise. Expert Opin Investig Drugs.
19:875–887. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yun SJ, Moon SK and Kim WJ:
Investigational cell cycle inhibitors in clinical trials for
bladder cancer. Expert Opin Investig Drugs. 22:369–377. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zaravinos A, Lambrou GI, Volanis D,
Delakas D and Spandidos DA: Spotlight on differentially expressed
genes in urinary bladder cancer. PLoS One. 6:e182552011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Davies B, Waxman J, Wasan H, Abel P,
Williams G, Krausz T, Neal D, Thomas D, Hanby A and Balkwill F:
Levels of matrix metalloproteases in bladder cancer correlate with
tumor grade and invasion. Cancer Res. 53:5365–5369. 1993.PubMed/NCBI
|
|
7
|
Lee SJ, Cho SC, Lee EJ, Kim S, Lee SB, Lim
JH, Choi YH, Kim WJ and Moon SK: Interleukin-20 promotes migration
of bladder cancer cells through extracellular signal-regulated
kinase (ERK)-mediated MMP-9 protein expression leading to nuclear
factor (NF-κB) activation by inducing the upregulation of p21(WAF1)
protein expression. J Biol Chem. 288:5539–5552. 2013.PubMed/NCBI
|
|
8
|
Sato H and Seiki M: Regulatory mechanism
of 92 kDa type IV collagenase gene expression which is associated
with invasiveness of tumor cells. Oncogene. 8:395–405.
1993.PubMed/NCBI
|
|
9
|
Moon SK, Cha BY and Kim CH: ERK1/2
mediates TNF-alpha-induced matrix metalloproteinase-9 expression in
human vascular smooth muscle cells via the regulation of NF-kappaB
and AP-1: involvement of the ras dependent pathway. J Cell Physiol.
198:417–427. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sato H, Kita M and Seiki M: v-Src
activates the expression of 92-kDa type IV collagenase gene through
the AP-1 site and the GT box homologous to retinoblastoma control
elements. A mechanism regulating gene expression independent of
that by inflammatory cytokines. J Biol Chem. 268:23460–23468.
1993.PubMed/NCBI
|
|
11
|
Kumar B, Koul S, Petersen J, Khandrika L,
Hwa JS, Meacham RB, Wilson S and Koul HK: p38 mitogen-activated
protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer
by modulation of MMP-2 and MMP-9 activity. Cancer Res. 70:832–841.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Krantz SB: Erythropoietin. Blood.
77:419–434. 1991.PubMed/NCBI
|
|
13
|
Ebert BL and Bunn HF: Regulation of the
erythropoietin gene. Blood. 94:1864–1877. 1999.PubMed/NCBI
|
|
14
|
Richmond TD, Chohan M and Barber DL:
Turning cells red: signal transduction mediated by erythropoietin.
Trends Cell Biol. 15:146–155. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Westenfelder C and Baranowski RL:
Erythropoietin stimulates proliferation of human renal carcinoma
cells. Kidney Int. 58:647–657. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lester RD, Jo M, Campana WM and Gonias SL:
Erythropoietin promotes MCF-7 breast cancer cell migration by an
ERK/mitogen-activated protein kinase-dependent pathway and is
primarily responsible for the increase in migration observed in
hypoxia. J Biol Chem. 280:39273–39277. 2005. View Article : Google Scholar
|
|
17
|
Mohyeldin A, Lu H, Dalgard C, Lai SY,
Cohen N, Acs G and Verma A: Erythropoietin signaling promotes
invasiveness of human head and neck squamous cell carcinoma.
Neoplasia. 7:537–543. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fu P, Jiang X and Arcasoy MO:
Constitutively active erythropoietin receptor expression in breast
cancer cells promotes cellular proliferation and migration through
a MAP-kinase dependent pathway. Biochem Biophys Res Commun.
379:696–701. 2009. View Article : Google Scholar
|
|
19
|
Paragh G, Kumar SM, Rakosy Z, Choi SC, Xu
X and Acs G: RNA interference-mediated inhibition of erythropoietin
receptor expression suppresses tumor growth and invasiveness in
A2780 human ovarian carcinoma cells. Am J Pathol. 174:1504–1514.
2009. View Article : Google Scholar
|
|
20
|
Belda-Iniesta C, Perona R, de Carpeno JC,
Cejas P, Casado E, Manguan-Garcia C, Ibanez de Caceres I,
Sanchez-Perez I, Andreu FB, Ferreira JA, Aguilera A, Dela PJ,
Perez-Sanchez E, Madero R, Feliu J, Sereno M and Gonzalez-Baron M:
Human recombinant erythropoietin does not promote cancer growth in
presence of functional receptors expressed in cancer cells. Cancer
Biol Ther. 6:1600–1605. 2007. View Article : Google Scholar
|
|
21
|
Liu WM, Powles T, Shamash J, Propper D,
Oliver T and Joel S: Effect of haemopoietic growth factors on
cancer cell lines and their role in chemosensitivity. Oncogene.
23:981–990. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
LaMontagne KR, Butler J, Marshall DJ,
Tullai J, Gechtman Z, Hall C, Meshaw A and Farrell FX: Recombinant
epoetins do not stimulate tumor growth in erythropoietin
receptor-positive breast carcinoma models. Mol Cancer Ther.
5:347–355. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee SJ, Lee EJ, Kim SK, Jeong P, Cho YH,
Yun SJ, Kim S, Kim GY, Choi YH, Cha EJ, Kim WJ and Moon SK:
Identification of pro-inflammatory cytokines associated with muscle
invasive bladder cancer; the roles of IL-5, IL-20, and IL-28A. PLoS
One. 7:e402672012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moon SK, Kim HM, Lee YC and Kim CH:
Disialoganglioside (GD3) synthase gene expression suppresses
vascular smooth muscle cell responses via the inhibition of ERK1/2
phosphorylation, cell cycle progression, and matrix
metalloproteinase-9 expression. J Biol Chem. 279:33063–33070. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Besson A, Dowdy SF and Roberts JM: CDK
inhibitors: cell cycle regulators and beyond. Dev Cell. 14:159–169.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fang J, Menon M, Kapelle W, Bogacheva O,
Bogachev O, Houde E, Browne S, Sathyanarayana P and Wojchowski DM:
EPO modulation of cell-cycle regulatory genes, and cell division,
in primary bone marrow erythroblasts. Blood. 110:2361–2370. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Panzenböck B, Bartunek P, Mapara MY and
Zenke M: Growth and differentiation of human stem cell
factor/erythropoietin-dependent erythroid progenitor cells in
vitro. Blood. 92:3658–3668. 1998.PubMed/NCBI
|
|
29
|
Stein JP, Ginsberg DA, Grossfeld GD,
Chatterjee SJ, Esrig D, Dickinson MG, Groshen S, Taylor CR, Jones
PA, Skinner DG and Cote RJ: Effect of p21WAF1/CIP1 expression on
tumor progression in bladder cancer. J Natl Cancer Inst.
90:1072–1079. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sharia SF, Kim J, Raptidis G, Ayala GE and
Lerner SP: Association of p53 and p21 expression with clinical
outcome in patients with carcinoma in situ of the urinary bladder.
Urology. 61:1140–1145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cordon-Cardo C: Mutations of cell cycle
regulators. Biological and clinical implications for human
neoplasia. Am J Pathol. 147:545–560. 1995.PubMed/NCBI
|