Introduction
Osteosarcoma (OS) is the most common primary bone
malignancy and the eighth most common type of cancer among
children, comprising 2.4% of all malignancies in pediatric patients
and ~35% of all bone cancers (1).
The overall incidence is five cases per million individuals per
year (1). OS is a devastating
disease, characterized by high local aggressiveness and a tendency
to metastasize to the lungs and distant bones. Pulmonary metastasis
occurs in ~40–50% of OS patients and remains a major cause of fatal
outcome (2–4). The cure rate of OS is ~65% for
patients with localized diseases. When presenting with metastases
at the time of diagnosis, the survival rate is 25% (4,6).
Despite modern multidisciplinary treatments including chemotherapy
and surgery, the 5-year survival rate of osteosarcoma patients
remains 60–70% (1). Hence, there is
an urgent need to develop novel approaches to treat osteosarcoma
patients, particularly, to identify and confirm potential
therapeutic targets involved in OS development and progression.
A new approach to therapeutic strategy is emerging,
based on the peculiar metabolism of the cancer cell. Specifically,
glycolysis has long been considered the main source of energy for
the cancer cell (7).
Fructose-bisphosphate aldolase (EC 4.1.2.13) is involved in
glycolysis by converting fructose 1,6-diphosphate into
dihydroxyacetone phosphate and glyceraldehyde-3-phosphate (8). The three aldolase isozymes (A, B and
C) have a tetramer structure with identical molecular weights of
~160 kDa. It is well known that cancer cells with a high glycolytic
rate often exhibit an aberrant expression of all glycolytic enzymes
(8). It has been found that the
control of glycolysis in rapidly growing tumor cells occurs at
least partly at the level of the so-called consuming block (from
aldolase to lactate dehydrogenase) (9). Accumulation of
fructose-1,6-bisphosphate resulting from inhibition of
aldolase-catalyzed cleavage should stop glycolysis and, therefore,
cancer development and progression (8).
A recent study revealed that the expression of
aldolase A (ALDOA) was significantly higher in OS patients with
shorter survival time, suggesting that ALDOA is a negative survival
marker of OS and may be implicated in OS development and
progression (10). In the present
study, for the first time we assessed the functional role of ALDOA
in OS cell invasion and survival in vitro and in
vivo, using human OS cell lines and an orthotopic xenograft OS
nude mouse model.
Materials and methods
Cells lines, plasmids, reagents and
mice
MG-63 and U-2 human OS cell lines were purchased
from the American Type Culture Collection (ATCC; Rockville, MD,
USA). Human ALDOA cDNA was subcloned into the pcDNA 3.1
expression vector (11).
ALDOA (sc-29664-V) shRNA lentiviral particles, control shRNA
lentiviral particles-A (sc-108080), and anti-ALDOA (N-15)
(sc-12059) and anti-matrix metalloproteinase-2 (MMP-2) antibodies
(sc-53630) were purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). The aldolase activity in cell extracts was analyzed
with an aldolase test kit (CALD 015) purchased from Caldon Biotech
(Vista, CA, USA). DeadEnd™ Fluorometric TUNEL system was purchased
from Promega (Madison, WI, USA). ApopTag® Peroxidase In
Situ Apoptosis Detection kit (S7100) was purchased from Millipore
(Billerica, MA, USA). SuperFect™ transfection reagent was purchased
from Qiagen (Valencia, CA, USA). Puromycin, G418, cisplatin and all
chemicals of reagent grade were purchased from Sigma (St. Louis,
MO, USA). Five-week-old BALB/C female nude mice were purchased from
Central South University (Changsha, China) and were housed at the
Xiangya Hospital BioResources Centre. All animal care, breeding and
testing procedures were approved by the Laboratory Animal Users
Committee of Xiangya Hospital, Central South University, Changsha,
China.
Transfection and lentiviral
transduction
The human ALDOA expression constructs were
transfected into MG-63 cells using SuperFect™ transfection reagent,
and pools of stable transductants were generated via selection with
G418 (800 μg/ml) according to the manufacturer’s protocol (Qiagen).
Lentiviral transduction was performed in U-2 cells, and pools of
stable transductants were generated via selection with puromycin (5
μg/ml) according to the manufacturer’s protocol (Santa Cruz
Biotechnology).
In vitro cell invasion assay
Transwell® cell invasion assays (Corning
Life Sciences, Tewksbury, MA, USA) were performed as previously
described (12). Briefly,
Transwell® cell-culture chambers with an 8-μm pore size
(BD Biosciences, Bedford, MA, USA) for 24-well plates were coated
with 50 μl Matrigel (BD Biosciences). OS cells were seeded into the
upper chamber at 5×105 cells/well in RPMI-1640
serum-free medium. Complete medium (600 ml) was added to the lower
chamber. Cells were allowed to invade for 24 h followed by fixation
and staining with crystal violet. Invaded cells which adhered to
the bottom of the filter were counted in 10 random fields per
chamber under a microscope. Each experiment was repeated three
times in triplicates.
Western blot analysis
Immunoblotting was performed with the respective
antibodies. Briefly, cells were dissolved in 250 μl of 2X SDS
loading buffer (62.5 mm Tris-HCl, pH 6.8, 2% SDS, 25% glycerol,
0.01% bromophenol blue, 5% 2-mercaptoethanol), and incubated at
95°C for 10 min. Equal amounts of proteins for each sample were
separated by 10% SDS-polyacrylamide gel and blotted onto a
polyvinylidene difluoride microporous membrane (Millipore).
Membranes were incubated for 1 h with a 1/1,000 dilution of primary
antibody, and then washed and revealed using secondary antibodies
with horseradish peroxidase conjugate (1/5,000, 1 h). Peroxidase
was revealed with a GE Healthcare ECL kit. Proteins were quantified
before being loaded onto the gel.
Measurement of apoptosis by terminal
deoxynucleotidyl transferase mediated nick-end labeling (TUNEL)
assay
The TUNEL assay was performed using the DeadEnd™
Fluorometric TUNEL system according to the manufacturer’s protocol
(Promega). Cells were treated with cisplatin (10 nM) for 8 h.
Apoptotic cells exhibit a strong nuclear green fluorescence that
can be detected using a standard fluorescein filter. All cells
stained with DAPI exhibit a strong blue nuclear fluorescence. The
slides were observed using fluorescence microscopy and the relative
apoptotic cells were determined by counting TUNEL-positive cells in
five random fields (magnification, ×100) for each sample.
Establishment of an orthotopic xenograft
OS nude mouse model
OS cells were mixed with 50% Matrigel to a
concentration of 2×106 cells/ml. Mice were anesthetized
by intraperitoneal injection of ketamine (100 mg/kg body weight)
and xylazine (10 mg/kg body weight). A volume of 10 μl of
cells/Matrigel solution was injected into the left tibia of
individual nude mice using a 27-gauge needle (12). The needle was inserted into the
tibial tuberosity and advanced using a drilling motion to avoid
fracture of the bone. The mice were monitored three times weekly
for tumor growth and signs of distress. Tumors were measured in the
anteroposterior and lateral planes using digital callipers. Leg
volume and tumor volume were calculated using the formula: 4/3π
(1/4(AP + L)2 where AP is the anteroposterior
measurement and L is the lateral measurement (13). The volume of the contralateral limb
was subtracted from the tumor-bearing limb to calculate the actual
tumor volume. Mice were weighed using digital scales. Tumor growth
was evaluated until death or sacrifice when tumor dimensions
exceeded 5% of the body weight or mice showed dyspnea, abnormal
posture, >20% body weight loss, difficulty with ambulation or
any other clinical sign of metastatic disease causing significant
pain or distress, according to the institutional guidelines.
Clonogenic lung metastasis assay
Clonogenic lung metastasis assays were performed as
previously described (14,15). Briefly, lungs from each individual
animal were minced into 1-mm pieces, and digested with 5 ml enzyme
cocktail containing 1 mg/ml collagenase IV and 6 units/ml elastase
in PBS for 1 h at 4°C with rotation. Cell suspensions were filtered
through 70-Amnylon cell strainers and washed two times with Hank’s
buffered saline, and then resuspended in complete medium. The cells
were then cultured in 10-cm tissue culture dishes and treated with
1.25 mg/ml of G418 or 5 μg/ml of puromycin to allow only the growth
of MG-63 and U-2 cells, respectively. When colonies of the growing
cells became visible (8–14 days), the plates were washed with
phosphate-buffered saline, fixed with methanol and stained with
crystal violet. The colonies were counted independently by two
investigators, blinded to the group to which each nude mouse
belonged, and the total colony number/lungs was calculated for each
animal.
Immunohistochemistry
Apoptosis in the primary OS tumor tissue was
evaluated using the ApopTag® Peroxidase In Situ
Apoptosis Detection kit according to the manufacturer’s protocol
(Millipore). Light hematoxylin was used for counterstaining. After
staining, apoptosis was determined from 500 randomly selected cells
as the proportion of cells with apoptotic nuclei.
Statistical analysis
Statistical analyses were performed with SPSS for
Windows 10.0. Data values are expressed as means ± SD. Comparison
of the means between two independent groups was performed with the
Student’s t-test. Comparison of the means among multiple groups was
performed with one-way ANOVA followed by post hoc pairwise
comparisons using Tukey’s tests. Two-tailed P<0.05 was
considered to indicate a statistically significant result in the
present study.
Results
As shown in Fig. 1, while ALDOA was amply
expressed in U-2 OS cells, it was expressed at a relatively low
constitutive level in the MG-63 cells. The two cell lines allowed
the specific ALDOA overexpression or knockdown studies to be
performed in the context of the study goals. Thus, we stably
transfected MG-63 cells with an ALDOA expression vector to
overexpress ALDOA, and we stably transduced U-2 cells with
ALDOA-shRNA to knock down ALDOA. Compared with the controls, ALDOA
was overexpressed ~2-fold in the MG-63 cells, and the endogenous
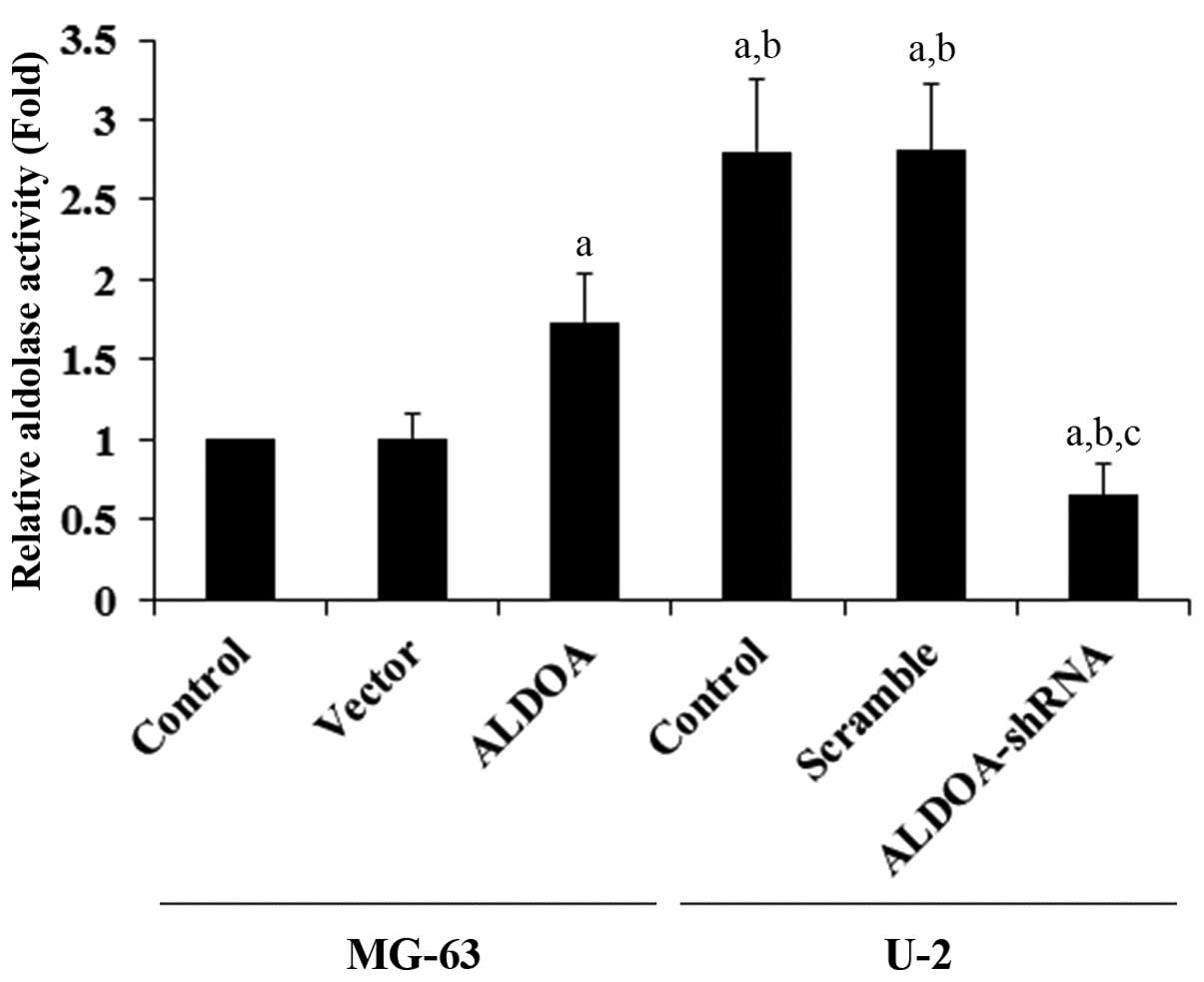
ALDOA level was knocked down ~80% in the U-2 cells (Fig. 1). Aldolase activity assays using the
cell extracts showed that U-2 cells had higher constitutive
aldolase activity when compared with that in the MG-63 cells
(Fig. 2). ALDOA overexpression
significantly increased the aldolase activity in the MG-63 cells,
when compared with the controls. On the other hand, ALDOA knockdown
markedly decreased the aldolase activity in the U-2 cells (Fig. 2).
 | Figure 1Aldolase A (ALDOA) expression in
osteosarcoma cells with overexpression or knockdown of ALDOA. (A)
In the MG-63 cells, expression of ALDOA in the control cells
(control, lane 1), cells stably transfected with the empty pcDNA3
vector (vector, lane 2), and cells stably transfected with ALDOA
(lane 3) was analyzed by western blot analysis. In the U-2 cells,
expression of ALDOA in control cells (control, lane 4), cells
stably transduced with scramble control shRNA (scramble, lane 5),
and cells stably transduced with ALDOA-shRNA (lane 6) was analyzed
by western blot analysis. β-actin blotting was used as a loading
control. Protein blots were measured by densitometry. (B) Density
of the ALDOA blot was normalized against that of β-actin to obtain
a relative blot density, which was expressed as fold-change to the
relative ALDOA blot density of MG-63 control cells (designated as
1). aP<0.05 compared with (MG-63) control and vector;
bP<0.05 compared with (MG-63) ALDOA;
cP<0.05 compared with (U-2) control and scramble. |
To examine the effect of ALDOA on OS cell invasion,
we performed in vitro cell invasion assays and examined the
MMP expression level in OS cells. Overexpression of ALDOA in MG-63
cells increased cell invasion by ~1-fold when compared with that of
the controls, while knockdown of ALDOA in U-2 cells decreased cell
invasion by ~80% (Fig. 3). A
similar trend in data was observed for the expression of MMP-2
(Fig. 4).
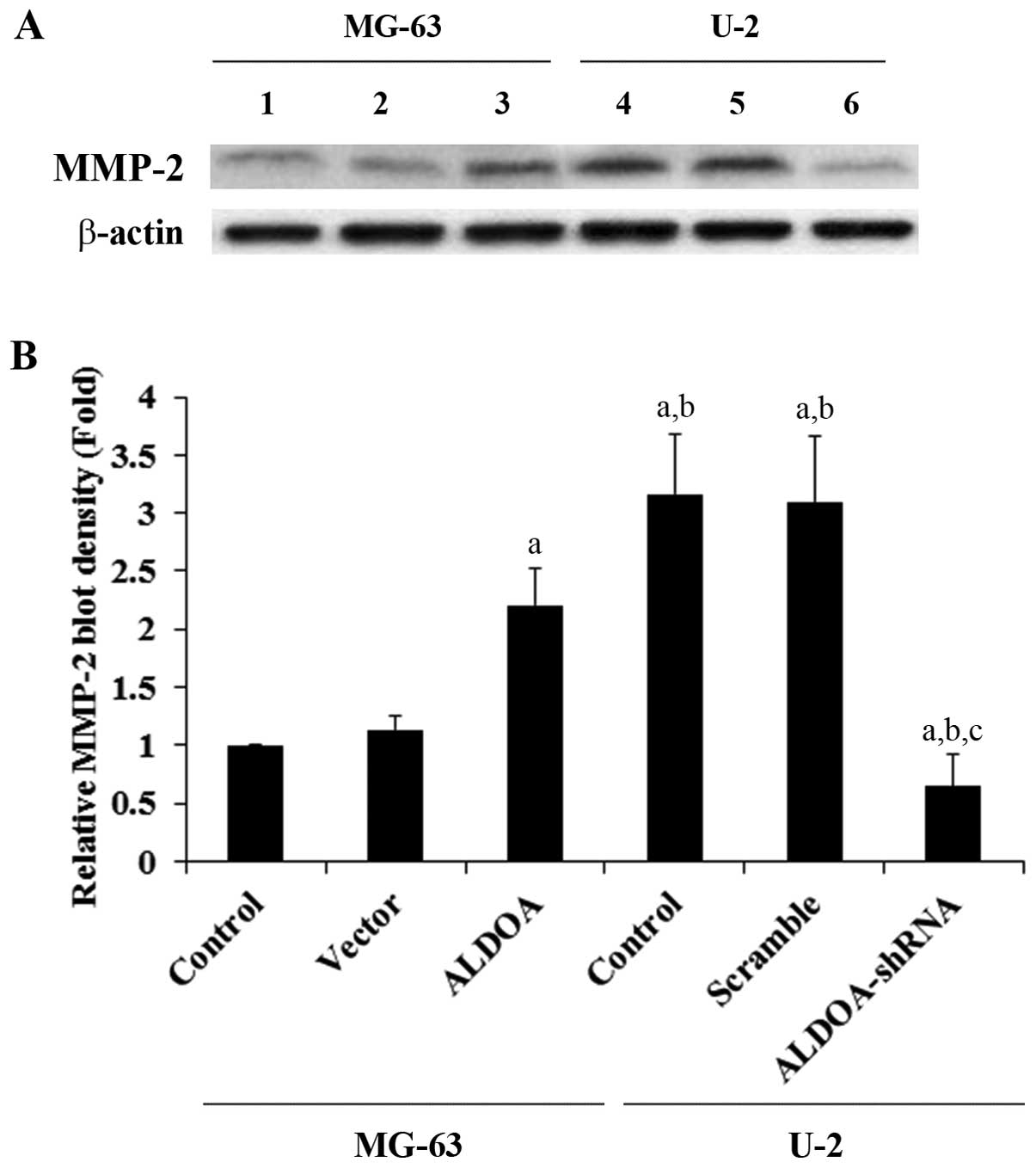 | Figure 4Matrix metalloproteinase-2 (MMP-2)
expression in osteosarcoma cells with overexpression or knockdown
of aldolase A (ALDOA). (A) In MG-63 cells, expression of MMP-2 in
control cells (control, lane 1), cells stably transfected with
empty pcDNA3 vector (vector, lane 2), and cells stably transfected
with ALDOA (lane 3) was analyzed with western blot analysis. In U-2
cells, expression of MMP-2 in control cells (control, lane 4),
cells stably transduced with scramble control shRNA (scramble, lane
5), and cells stably transduced with ALDOA-shRNA (lane 6) was
analyzed with western blot analysis. β-actin blotting was used as a
loading control. Protein blots were measured by densitometry. (B)
Density of the MMP-2 blot was normalized against that of β-actin to
obtain a relative blot density, which was expressed as fold-change
to the relative MMP-2 blot density of MG-63 control cells
(designated as 1). aP<0.05 compared with (MG-63)
control and vector; bP<0.05 compared with (MG-63)
ALDOA; cP<0.05 compared with (U-2) control and
scramble. |
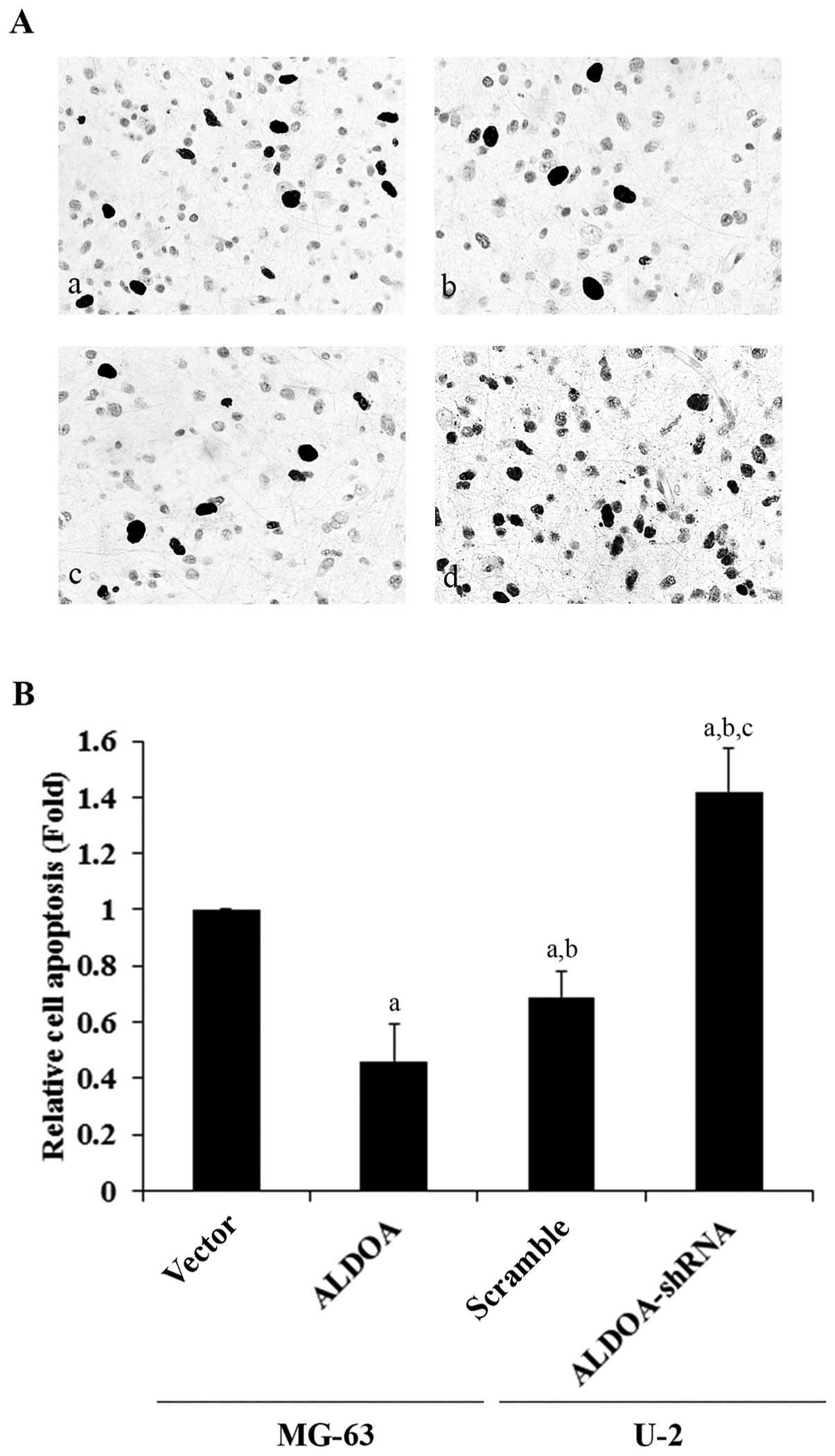
To explore the effect of ALDOA on OS survival, we
examined cell apoptosis in OS cells treated with 10 nM of
cisplatin, an apoptosis-inducing chemotherapeutic agent commonly
used to treat OS. Overexpression or knockdown of ALDOA did not
significantly alter the rate of cell apoptosis in both the MG-63
and U-2 cells under normal culture conditions (Fig. 5A). However, in the MG-63 cells
treated with cisplatin, overexpression of ALDOA significantly
decreased the rate of cell apoptosis when compared with the
controls (Fig. 5B). In the U-2
cells, knockdown of ALDOA significantly increased cell apoptosis in
the presence of cisplatin (Fig.
5C).
To assess the role of ALDOA in OS progression and
metastasis in vivo, we employed an orthotopic xenograft OS
nude mouse model. MG-63 cells stably transfected with the empty
pcDNA3 expression vector or ALDOA, and U-2 cells stably transduced
with scramble control shRNA or ALDOA-shRNA were used for
intra-tibial injection, respectively. Twenty-eight days after the
injection, 67% (6/9) of the mice in the vector control (vector)
group, 100% (9/9) in the ALDOA overexpression group, 100% (9/9) in
the scramble control (scramble) group, and 22% (2/9) in the ALDOA
knockdown (ALDOA-shRNA) group exhibited obvious dyspnea and
distress. All animals were sacrificed on day 28 post injection, and
the lungs were collected. As shown in Fig. 6, mice injected with the MG-63 cells
overexpressing ALDOA showed significantly larger primary tumors
than those noted in the vector controls at day 21 post injection.
In contrast, mice injected with the U-2 cells with ALDOA knockdown
showed significantly smaller primary tumors than those noted in the
scramble controls at day 14 post-injection. Compared with the
vector control group, the ALDOA overexpression group showed
obviously more metastatic nodules on the lung surface as well as
more swollen and congested lungs (Fig.
7A). In comparison with the scramble control group, the ALDOA
knockdown group showed apparently fewer metastatic nodules on the
lung surface as well as less swollen and congested lungs (Fig. 7A). To quantitate the pulmonary
metastasis, clonogenic lung metastasis assays were performed. As
shown in Fig. 7B, the number of
lung metastases in the ALDOA overexpression group were
significantly more than these numbers in the vector control group.
In contrast, the ALDOA knockdown group showed markedly fewer
metastases than the scramble control group (Fig. 7C).
To explore the effect of ALDOA on OS cell survival
in vivo, we examined cell apoptosis in the primary tumors in
the orthotopic xenograft OS mouse nude model. As shown in Fig. 8, mice injected with the MG-63 cells
overexpressing ALDOA showed significantly lower OS cell apoptosis
rates in the primary tumors than those injected with the vector
control cells. On the other hand, mice injected with the U-2 cells
with knockdown of ALDOA showed significantly higher OS cell
apoptosis rates in the primary tumors than those injected with the
scramble control cells.
Discussion
Inhibiting cancer cell glycolysis is an emerging
therapeutic strategy for cancer (8). A recent study suggested that ALDOA, an
important enzyme involved in glycolysis (8), is a negative survival marker of OS and
may be implicated in OS development and progression. In the present
study, our in vitro data revealed that ALDOA promoted OS
cell invasion and survival, and our in vivo data
demonstrated an important role of ALDOA in promoting OS tumor
growth and metastasis.
MG-63 and U-2 cells were used as OS cell models in
the present study. MG-63 cells expressed a relatively low
constitutive level of ALDOA when compared with the U-2 cells. Thus,
overexpression and knockdown of ALDOA were respectively performed
in the two cell lines to approach the study objectives from
different angles. Changes in the aldolase activity were in line
with those in the ALDOA expression levels, indicating that
overexpression and knockdown of ALDOA indeed led to an alteration
in the enzymatic activity in the OS cells.
OS is characterized by high local aggressiveness and
a tendency to metastasize to the lungs, which remains a major cause
of fatal outcome (4). Thus, we
performed in vitro cell invasion assays to explore the
effect of ALDOA on OS cell invasiveness. Overexpression of ALDOA
increased cell invasion in MG-63 cells, which had a relatively low
constitutive level of ALDOA expression and invasive activity, while
knockdown of ALDOA nearly abolished cell invasion in the U-2 cells,
which had a relatively high constitutive level of ALDOA expression
and invasive activity. The findings suggest that ALDOA is critical
for OS cell invasion. Among the different MMPs, MMP-2 showed
expression level changes in line with the cell invasive activity
changes in the OS cells, suggesting that ALDOA promotes OS cell
invasion through upregulation of MMP-2 expression. Further studies
are needed to ascertain how ALDOA regulates MMP-2 expression.
Cell survival against apoptotic stress is critical
for cancer progression and metastasis (16). In the present study, we used a
relatively small concentration of cisplatin (10 nM) to induce
apoptotic stress without killing most of the cells. In the presence
of cisplatin, overexpression of ALDOA in the MG-63 cells
significantly decreased cell apoptosis, while knockdown of ALDOA in
the U-2 cells markedly increased cell apoptosis compared with the
controls. The findings indicate that ALDOA is important for OS cell
survival against apoptotic stress, which not only suggests a
functional role for ALDOA in OS progression and metastasis, but
also implicates ALDOA in the development of OS chemoresistance.
Cisplatin elicits DNA repair mechanisms by crosslinking DNA, which
in turn activates apoptosis when repair proves impossible (17). It is still unclear whether ALDOA
impacts OS cell survival against other types of chemotherapeutic
agents. Further studies with more types of chemotherapeutic agents
and OS cell lines would elucidate this issue.
Based on the in vitro evidence that ALDOA
plays an important role in OS cell invasion and survival, we used
an orthotopic xenograft OS nude mouse model to further explore the
functional role of ALDOA in OS progression and metastasis in
vivo. As pulmonary metastasis is a major cause of fatal outcome
in OS, we focused on lung metastasis in the mouse model, and the
intra-tibial injection model in nude mice has proved to be a
biologically relevant and adequate animal model for the induction
of reproducible pulmonary metastasis (15). A combination of clinical signs,
organ examinations, tumor volume analyses, and quantitative lung
metastasis assays in the animal model demonstrated that ALDOA
promotes OS primary tumor growth and pulmonary metastasis in
vivo. Additionally, overexpression and knockdown of ALDOA
decreased and increased cell apoptosis in the primary OS tumors,
respectively, confirming the in vitro promoting effects of
ALDOA on OS cell survival against the apoptotic stress induced by
low-dose cisplatin.
The aldolase (ALDO) isozymes (A, B and C) are
encoded by three different genes, differentially expressed during
development. ALDOA is mainly produced by the developing embryo and
in adult muscle; ALDOB is produced by liver, kidney and intestine;
and ALDOC is mainly produced by brain and other nervous tissue.
ALDOA and ALDOB have been associated with poor prognosis of OS and
hepatocarcinomas, respectively (10,18).
Future studies of whether and how ALDOB and ALDOC are involved in
OS development and progression are warranted.
In conclusion, the present study provides the first
in vitro and in vivo evidence supporting a critical
functional role of ALDOA in OS progression and metastasis,
suggesting that ALDOA could serve as a novel therapeutic target in
OS. Additionally, our results also suggest that ALDOA is involved
in the development of OS chemoresistance.
References
|
1
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2010. View Article : Google Scholar
|
|
2
|
Bacci G, Briccoli A, Rocca M, et al:
Neoadjuvant chemotherapy for OS of the extremities with metastases
at presentation: recent experience at the Rizzoli Institute in 57
patients treated with cisplatin, doxorubicin, and a high dose of
methotrexate and ifosfamide. Ann Oncol. 14:1126–1134. 2003.
View Article : Google Scholar
|
|
3
|
Kager L, Zoubek A, Potschger U, et al:
Primary metastatic OS: presentation and outcome of patients treated
on neoadjuvant Cooperative OS Study Group protocols. J Clin Oncol.
21:2011–2018. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ta HT, Dass CR, Choong PF and Dunstan DE:
Osteosarcoma treatment: state of the art. Cancer Metastasis Rev.
28:247–263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gorlick R, Anderson P and Andrulis I:
Biology of childhood osteogenic sarcoma and potential targets for
therapeutic development: meeting summary. Clin Cancer Res.
9:5442–5453. 2003.PubMed/NCBI
|
|
6
|
Wittig JC, Bickels J and Priebat D:
Osteosarcoma: a multidisciplinary approach to diagnosis and
treatment. Am Fam Physician. 65:1123–1132. 2002.PubMed/NCBI
|
|
7
|
Gatenby RA and Gillies RJ: Why do cancers
have high aerobic glycolysis? Nat Rev Cancer. 4:891–899. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scatena R, Bottoni P, Pontoglio A,
Mastrototaro L and Giardina B: Glycolytic enzyme inhibitors in
cancer treatment. Expert Opin Investig Drugs. 17:1533–1545. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marin-Hernandez A, Rodríguez-Enríquez S,
Vital-González PA, et al: Determining and understanding the control
of glycolysis in fast-growth tumor cells. Flux control by an
over-expressed but strongly product-inhibited hexokinase. FEBS J.
273:1975–1988. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen X, Yang TT, Zhou Y, et al: Proteomic
profiling of osteosarcoma cells identifies aldoa and sult1a3 as
negative survival markers of human osteosarcoma. Mol Carcinog. Sep
4–2012.(Epub ahead of print).
|
|
11
|
Sakakibara M, Takahashi I, Takasaki Y,
Mukai T and Hori K: Construction and expression of human aldolase A
and B expression plasmids in Escherichia coli host. Biochim
Biophys Acta. 1007:334–342. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dass CR, Ek ET, Contreras KG and Choong
PF: A novel orthotopic murine model provides insights into cellular
and molecular characteristics contributing to human osteosarcoma.
Clin Exp Metastasis. 23:367–380. 2006. View Article : Google Scholar
|
|
13
|
Ek ETH, Dass CR, Contreras KG and Choong
PFM: Inhibition of orthotopic osteosarcoma growth and metastasis by
multitargeted antitumour activities of pigment epithelium-derived
factor. Clin Exp Metastasis. 24:93–106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pilones KA, Kawashima N, Yang AM, et al:
Invariant natural killer T cells regulate breast cancer response to
radiation and CTLA-4 blockade. Clin Cancer Res. 15:597–606. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Liao Q, Li K, et al: Knockdown of
endothelin A receptor expression inhibits osteosarcoma pulmonary
metastasis in an orthotopic xenograft mouse model. Mol Med Rep.
5:1391–1395. 2012.
|
|
16
|
Hopkin K, Edwards P, Harris A, Klausner R,
Peters G, Selby P and Stanley M: Cancer. Molecular Biology of the
Cell. Alberts B, Johnson A and Lewis J: 4th edition. Garland
Science; New York: pp. 1324–1325. 2002
|
|
17
|
Rosenberg B, Vancamp L, Trosko JE and
Mansour VH: Platinum compounds: a new class of potent antitumour
agents. Nature. 222:385–386. 1969. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peng SY, Lai PL, Pan HW, Hsiao LP and Hsu
HC: Aberrant expression of the glycolytic enzymes aldolase B and
type II hexokinase in hepatocellular carcinoma are predictive
markers for advanced stage, early recurrence and poor prognosis.
Oncol Rep. 19:1045–1053. 2008.
|