Introduction
Tamoxifen (TAM), a selective estrogen receptor
modulator (SERM), has been studied for over 20 years as an
anti-estrogenic drug used to treat estrogen receptor (ER)-positive
breast cancer. By competing for binding at the ER, TAM has been
proven to reduce the risk of developing breast cancer by 49%
through its antagonistic properties (1). Binding of TAM results in decreased
expression of genes that affect cell proliferation leading to
diminished cell divisions in breast cancer (2). However, in the endometrium, TAM has
been shown to act as a partial estrogen agonist, thus leading to
the inappropriate proliferation of endometrial tissues.
Consequently, the risk of developing endometrial cancer is
increased by 3-fold in TAM-treated women (3). In addition, the agonistic effect of
TAM has been shown to exhibit high variability depending upon cell
type, promoter context, ambient estradiol concentration and ER
subtype (α or β) (4,5). Furthermore, TAM has been shown to have
significant in vitro cytoxic and cytostatic effects on ER
(+) and ER (−) breast cancer cells (6,7). A
study by Reddel et al indicated that the effects of TAM are
dose- and ER status-dependent (8).
Specifically, their study showed that at low doses TAM effects are
ER-mediated, whereas at higher doses TAM effects appeared to be
ER-independent.
The antagonistic and agonistic properties inherent
to TAM are also present in its numerous metabolites, specifically
4′-hydroxy-tamoxifen (4-OH-TAM). With the addition of a hydroxyl
group, 4-OH-TAM has been shown to have a higher potency than TAM
both in vitro and in vivo corresponding to a higher
affinity for the ER (9). Of
interest is the effect of 4-OH-TAM in ER-negative tissues, such as
the rat (10) and mouse livers
(11), where TAM-DNA adducts and
4-OH-TAM-induced DNA adducts have been demonstrated. Moreover, a
study by Shibutani et al demonstrated TAM-DNA adduct
formation in ER-positive tissues, such as the human endometrium
(12). These results suggest the
possibility of an ER independent pathway for TAM and its
metabolites to induce cell proliferation.
A study by Castro-Rivera and Safe determined that
ER-positive HEC-1A human adenocarcinoma cells treated with TAM
exhibited both agonistic and antagonistic tendencies with respect
to cell proliferation depending on the concentration used (13). Another study by Perry et al
demonstrated the apopotic effects of TAM in breast cancer cells
regardless of ER status (14). The
ER status of HEC-1A and HEC-1B cells has been debated in the
literature. A recent study by Acconcia et al unequivocally
demonstrated the presence of ER-α in both cell lines (15). However, their immunofluoresence data
showed differences in the ER-α subcellular distribution, where
HEC-1A ER-α was observed in both the cytosol and the nucleus,
whereas, in HEC-1B cells ER-α was mostly located in the cytosol.
The aim of the present study was to investigate the in vitro
cytotoxicity of 4-OH-TAM and E2 in human endometrial
adenocarcinoma HEC-1A and HEC-1B cancer cell lines. We observed an
obvious decrease in HEC-1B and HEC-1A cell growth noted at 10 and
100 μM 4-OH-TAM and 100 μM E2. Furthermore, these
micromolar concentrations of 4-OH-TAM induced a non-apoptotic
cytotoxic effect.
Materials and methods
Cell lines and tissue culture
conditions
HEC-1B and HEC-1A human endometrial adenocarcinoma
cell lines were a generous gift from Dr. Cheryl Walker at M.D.
Anderson (Smithville, TX, USA). HEC-1B and HEC-1A cells were
maintained in MEM (cat. #11095-080) or McCoy’s 5A (cat. #11095-080)
purchased from Life Technologies (Grand Island, NY, USA)
respectively supplemented with 10% fetal bovine serum (FBS, cat.
#SH3039603) or charcoal stripped FBS (cat. #SH3006803; lacks
endogenous steroid hormones, CSFBS) (both purchased from Hyclone
Laboratories, Logan, UT, USA), 2% L-glutamine (cat. #G-7513) and 1%
sodium pyruvate (cat. #S8636) (both obtained from Sigma, St. Louis,
MO, USA). Cells were grown in 25 and 75 cm2 culture
flasks in a humidified atmosphere of 5% CO2 at 37°C.
Compounds
The compounds 4-OH-TAM (cat. #H-7904) and
E2 (cat. #E-2758) were purchased from Sigma and
dissolved in 100% ethanol, stored and protected from light in stock
solutions of 1 mM at −20°C. The final concentration of ethanol in
the culture media was consistently <0.1% (v/v).
Exposure to compounds and determination
of cell growth
The media corresponding to HEC-1B and HEC-1A cells
were replaced using 5% CSFBS 24 h prior to plating. Both cell lines
were seeded (10,000 cells/well) in triplicates into 96-well plates.
After 24 h, cells were incubated with 200 μl of various 4-OH-TAM or
E2 concentrations for 1–3 days at the conditions
described above. Cells grown in the absence of 4-OH-TAM and
E2 were used as a control. Media were changed on day 2
to ensure proper nutrient content and effective drug
concentration.
Cell viability was assessed using CellTiter
96®AQueous One Solution Cell Proliferation Assay (cat.
#G3580; Promega, Madison, WI, USA) every 24 h. Briefly, 20 μl of
3-(4,5-dimethyl-thiazol-2-yl)-5-(3-carboxymethoxphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(inner salt; MTS) and an electron-coupling reagent (phenazine
ethosulfate; PES) were added to each well. Plates were incubated
for 1–3 h at 37°C in a humidified 5% CO2 atmosphere.
Absorbance was read at 490 nm using an ELISA plate reader (BioRad,
Hercules, CA, USA). Statistical analysis was conducted using a
two-way ANOVA with post-hoc comparisons.
DNA fragmentation assay
DNA extracts were prepared from adherent and
detached HEC-1B and HEC-1A cells treated with 10 μM 4-OH-TAM or
E2 (in 5% CSFBS media) for 24 h or cells treated with 10
μM 4-OH-TAM or 50 μM E2 (in 5% CSFBS media) for 6 or 12
h at the incubating conditions described above. DNA extracts from
HEC-1B and HEC-1A cells grown in the absence of 4-OH-TAM or
E2 were used as a control. In brief, DNA was isolated
using the Wizard® Genomic DNA purification kit (cat.
#A1120; Promega) and DNA laddering analysis was performed using a
DNA Laddering Assay kit (cat. #660990; Cayman, Ann Arbor, MI, USA)
following the manufacture’s instructions. DNA fragments were then
separated on a 2% Agarose E-Gel® (cat. #G5018-02; Life
Technologies) and visualized with ethidium bromide under
ultraviolet light.
Western blot analysis
Caspases
HEC-1A and HEC-1B cells were seeded in 25
cm2 culture flasks and allowed to grow to 85%
confluency. Cells were harvested and protein extracts were prepared
from cells (adherent and detached cells) grown in the absence
(control) or presence of 1, 10 or 100 μM 4-OH-TAM or E2
(in 5% CSFBS media) for 24 h at the incubating conditions
previously described followed by western blot analysis. In brief,
whole cell lysates were prepared from log phase cells with 4X
sample buffer (40% v/v glycerol, 4% SDS, 0.5% w/v bromophenol blue
and 0.16 M Tris pH 7.0) plus 10% β-mercaptoethanol, subjected to
gel electrophoresis on precast 12% SDS-polyacrylamide gels (cat.
#456-1043; BioRad) and transferred to a polyvinyldene fluoride
(PVDF) membrane (cat. #IPVH304F0; Merck Millipore Ltd., Co. Cork,
Ireland). The membrane was blocked for 1 h with 5% milk in PBS and
probed with 0.25 μg/ml mouse anti-caspase-8 monoclonal antibody
(cat. #551242; BD Biosciences, San Diego, CA, USA) or with 2 μg/ml
mouse anti-caspase-3 monoclonal antibody (cat. #35-1600Z; Life
Technologies) for 1 h at room temperature with constant agitation
followed by a series of washes and incubated for 1 h with goat
anti-mouse HRP conjugate (1:3,000 dilution; cat. #170-6516;
BioRad). A chemiluminescent HRP signal detection system Amersham
ECL™ Prime Western Blotting Detection Reagent (cat. #RPN2232; GE
Healthcare, Buckinghamshire, UK) was used to detect the signal.
Growth inhibition vs. cell death
In order to differentiate between the cytostatic and
cytotoxic effects of 4-OH-TAM and E2, medium
corresponding to HEC-1B and HEC-1A cells was replaced using 5%
CSFBS 24 h prior to plating. Both cell lines were then seeded
(50,000 cells/well) in duplicates into 6-well plates. Cell counts
of two random wells were performed during plating to confirm
uniformity of cell distribution and to establish the actual initial
number of cells plated and 24 h later before drug exposure to
confirm the population doubling time (PDT). At this time, cells
were incubated with 2 ml of 10 μM 4-OH-TAM or 100 μM E2
for 24 h in 5% CSFBS media at the conditions previously described.
Cells grown in the absence of 4-OH-TAM or E2 were used
as a control. Cell viability was determined by trypan blue dye
exclusion assay.
Results
Determination of cell growth after
exposure to the compounds
HEC-1B and HEC-1A cells were exposed to varying
concentrations of 4-OH-TAM and E2 to determine the
effects of these compounds on cell survival.
When comparing the different 4-OH-TAM or
E2 concentrations (10–1000 nM) used to treat the HEC-1B
cells, no significant difference in the percent survival was
observed (p≤0.260; p≤0.755) (Fig. 1A
and B). However, as shown in Fig.
2A, HEC-1B cells exposed to higher concentrations (1–100 μM) of
4-OH-TAM showed a significant difference in the percent cell
survival between concentrations (p≤0.0001). Furthermore, at the
concentrations of 10 and 100 μM, a definitive decrease in the
percent survival was noted when compared to the untreated cells
(percent survival indicated by solid line and set to 100%). In
contrast, as shown in Fig. 2B,
HEC-1B cells treated with the same concentrations of E2
underwent an initial proliferative response observed after 24 h at
1 and 10 μM, followed by an apparent decrease in cell survival that
did not appear to be different from the non-treated cells.
Treatment with 100 μM E2 resulted in complete cell
death. Furthermore, a significant difference in percent survival
was noted between E2 concentrations (p≤0.0001).
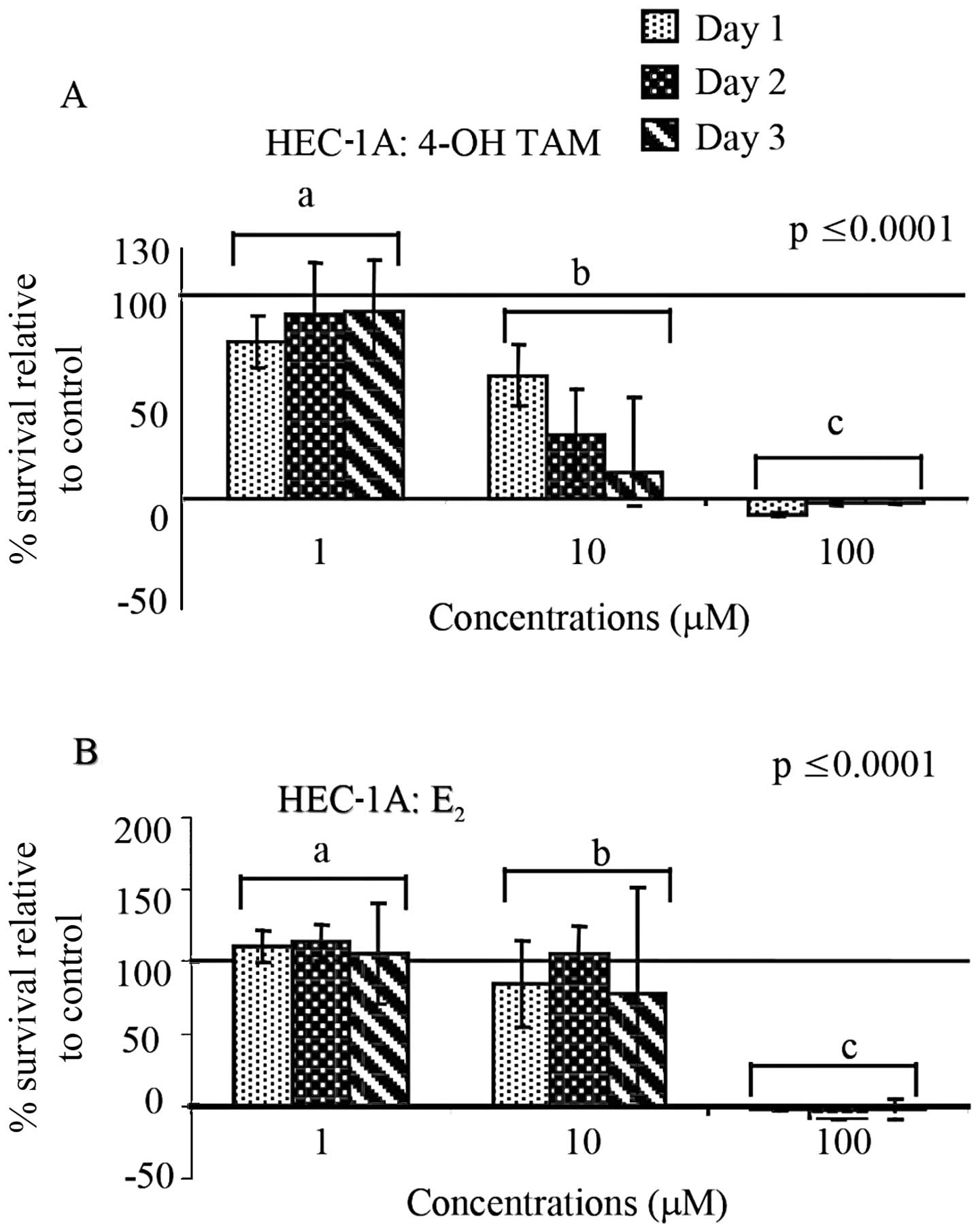
When HEC-1A cells were treated with the same high
concentrations (1–100 μM) as HEC-1B cells of 4-OH-TAM or
E2, we observed a significant difference in the
percentage of cell survival between concentrations (p≤0.0001,
similar to HEC-1B cells. When comparing the percent survival of
treated cells with the control, 4-OH-TAM appeared to decrease cell
survival at 10 μM and complete cell death was observed at 100 μM
(Fig. 3A). Similarly, as shown in
Fig. 3B, E2 caused
complete cell death at 100 μM and there were significant
differences between concentrations.
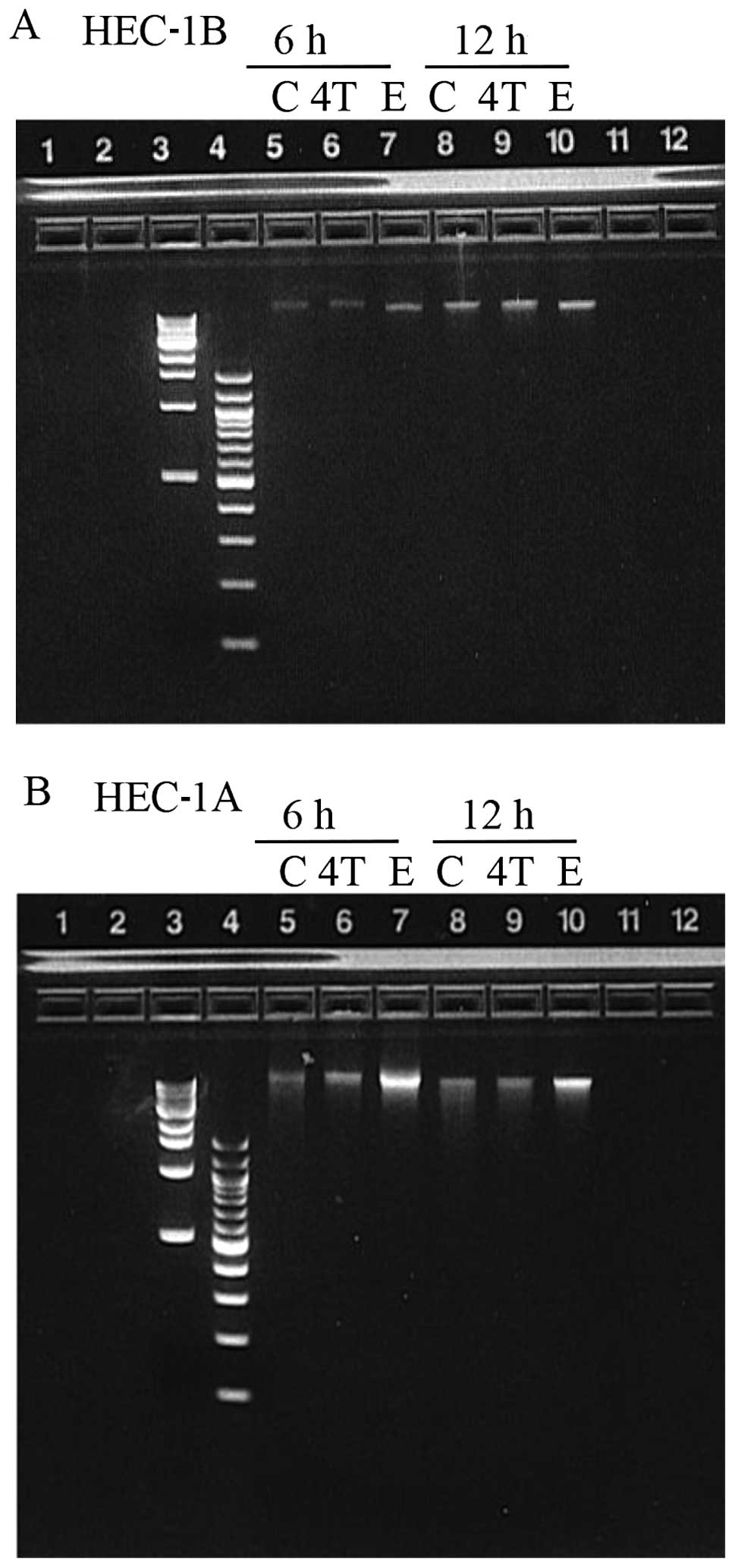
Absence of DNA laddering
To determine whether apoptosis was the underlying
cellular mechanism responsible for the decreased percent cell
survival observed in both cell lines exposed to 10 μM or 100 μM
4-OH-TAM or 100 μM E2 (when compared to the control);
DNA was extracted from adherent and floating HEC-1B and HEC-1A
cells after 24 h of exposure with 10 μM 4-OH-TAM or E2
and subjected to electrophoresis on a 2% agarose gel. No DNA
laddering was observed in any of the samples (data not shown). Due
to concern with the possibility of having extracted the DNA past a
time-point where DNA fragmentation could be observed, HEC-1B and
HEC-1A cells were incubated with 10 μM 4-OH-TAM or 50 μM
E2 and DNA was extracted at 6 and 12 h after exposure.
Cells were exposed to a higher E2 concentration (50 μM),
since we thought that 10 μM E2 was not high enough to
induce apoptosis (refer to Fig. 2B
and 3B). Again, no DNA laddering
was observed in any of the samples suggesting that apoptosis was
not the mechanism involved (Fig.
4).
Absence of caspase-3 and -8 activation as
determined by immunoanalysis
To determine whether activation of caspase-8 and -3
had occurred, western blot analysis of the protein extracts
collected from adherent and floating HEC-1B and HEC-1A cells grown
in the absence (control) or presence of 1 or 10 μM 4-OH-TAM or
E2 for 10 and 24 h was performed. We did not observed
the expected lower molecular weight bands for caspase-8 or -3 with
any of the concentrations used or time-points (data not shown).
Caspase-3 exists in cells as an inactive 32-kDa proenzyme and
during apoptosis pro-caspase-3 is cleaved into 17- and 12-kDa
active subunits by upstream proteases such as caspase-8. Similarly,
during apoptosis, the proform of caspase-8a 55-kDa protein, is
cleaved into smaller active subunits of 40/36 kDa (doublet) and 23
kDa.
Cytotoxic effect of 4-OH-TAM and
estradiol
Due to the absence of apoptotic markers, we wanted
to differentiate between the cytostatic and cytotoxic effects of
4-OH-TAM and E2. In order to do that cells were
incubated in the presence or absence of 10 μM 4-OH-TAM or 100 μM
E2 for 24 h in 5% CSFBS.
Trypan blue dye exclusion assay was used to assess
the cytotoxic effect of 4-OH-TAM or E2 (Fig. 5). Cell viability of HEC-1B and
HEC-1A cells treated with 10 μM 4-OH-TAM was determined to be
<25% in comparison to the number of living cells at the time of
exposure. When HEC-1B and HEC-1A cell lines were treated with 100
μM E2, 0% cell survival was observed. Untreated cells of
both cell lines exhibited proliferation of greater than 40% in
comparison to the number of living cells at the time of exposure.
These results confirmed that cell death occurred differentiating
4-OH-TAM and E2 cytotoxic effect from a cytostatic
effect.
Discussion
The present study examined the effects of 4-OH-TAM
and E2 on the proliferation of HEC-1B and HEC-1A human
endometrial adenocarcinoma cells. Since endometrial cell
proliferation is the most sensitive marker for differentiating
agonistic vs. antagonistic activity (16), endometrial cell lines were subjected
to hormonal treatments of 4-OH-TAM and E2 at varying
concentrations (nM to μM) for 1–3 days.
At concentrations ranging from 10–1000 nM of
4-OH-TAM or E2, HEC-1B cell proliferation did not differ
from that of the untreated cells. Due to the apparent lack of
effect of 4-OH-TAM on the growth patterns between treated and
untreated HEC-1B cells, we decided to increase the concentrations
of 4-OH-TAM and E2 to the micromolar range. When
subjecting HEC-1B and HEC-1A cells to 1 μM 4-OH-TAM for 1–3 days,
the growth patterns did not differ from the untreated cells, while
cells treated with 10 and 100 μM 4-OH-TAM underwent a significant
decrease in cell prolifieration when compared to the control. These
results differ from a study by Castro-Rivera and Safe (13) which determined that HEC-1A human
endometrial adenocarcinoma cells treated with 1 μM TAM underwent a
significant decrease in cell proliferation. When using 4-OH-TAM, a
decreased proliferation of HEC-1A cells was not observed until the
4-OH-TAM concentration was increased to 10 μM, thus suggesting a
difference in the effects of TAM and its metabolite 4-OH-TAM. This
is an interesting result considering 4-OH-TAM has been shown to
possess a higher potency than TAM (9).
While micromolar concentrations of 4-OH-TAM and
E2 appear to be clinically irrelevant, the plasma
concentration range of TAM measured in chemotherapy patients has
been recorded as 0.1–10 μM (17).
Furthermore, patients treated with higher doses of TAM (720 mg per
day) have serum levels of up to 3.5 μM with accumulated levels in
tissues reaching 16–30 times higher than that of the serum
(14). Therefore, the
concentrations of 4-OH-TAM utilized in this study mirror actual
concentrations of TAM recorded in patients undergoing treatment for
breast cancer, thus verifying the application of our chosen
concentrations.
Following observation of the HEC-1B and HEC-1A cells
exposed to 1 or 10 μM E2 for 1–3 days, the growth
patterns did not appear to be different from the untreated cells,
whereas 100 μM E2 caused an obvious decrease in cell
proliferation in both cell lines. Since HEC-1A and HEC-1B cells
contain ER-α (15), estrogen
treatment was hypothesized to induce an increase in cell growth in
comparison to the untreated cells. However, the concentrations of
estadiol used in this study were higher than physiological plasma
concentrations of estrogen (~1 nM) (18) and thus may have led to the cytotoxic
effect.
In order to determine the mechanism behind the
observed decrease in cell proliferation, we looked for DNA
laddering 24 h after exposure of the cells to 10 μM 4-OH-TAM or
E2. DNA gel electrophoresis failed to show DNA-laddering
in any of the samples. We were concerned with the possibility of
having extracted the DNA at a time-point when it was too late to
observe DNA fragmentation. Therefore, we extracted DNA at 6 and 12
h after exposure to 10 μM 4-OH-TAM or 50 μM E2. Another
concern was that the E2 concentration (10 μM) may not
have been high enough to induce apoptosis. Therefore, we increased
the E2 concentration to 50 μM for the 6 and 12 h
exposures. Neither of these measures resulted in the evidence of
apoptosis through DNA laddering. Similarly, Dietze et al
found that 1 μM TAM, but not an equimolar concentration of
4-OH-TAM, induced apoptosis in ER-positive normal human mammary
epithelial cells (HMECs) (19).
This suggests that 4-OH-TAM induces cell death through a mechanism
other than apoptosis.
Furthermore, western blot analysis of caspase-8 and
-3 expression failed to unequivocally show caspase activation. The
inactive form of caspase-8, a 55-kDa protein, is cleaved into
smaller subunits of 40/36 kDa (doublet) and 23 kDa upon activation,
whereas procaspase 3 (32 kDa) is cleaved into active 17- and 12-kDa
subunits. Our data excluded the death receptor pathway of the
apoptotic mechanism (which utilizes caspase-8) as the underlying
cellular mechanism of the observed cell death. Therefore, the
results of the present study and the lack of DNA laddering suggest
that apoptosis was not the underlying mechanism used to cause cell
death in the 4-OH-TAM and E2-treated HEC-1B and HEC-1A
cells.
Although apoptosis is the most common form of
programmed cell death there are alternative non-apoptotic
mechanisms that can lead to regulated cell death such as autophagy,
necroptosis or the non-regulated pathway of necrosis. In fact,
mammalian cells have been shown to express cell death proteases
even when they are not undergoing apoptosis (20). Furthermore, characteristics assumed
to be unique to apoptosis, such as chromatin condensation and even
DNA fragmentation, may not be strictly indicative of apoptosis
(21). On a kinetic scale,
morphological changes in cell structure may occur over a wide time
range depending on cell type as noted in hepatocytes (2–3 h) and
lymphocytes (36–48 h) (22).
Considering all the data from these studies, a form of cross-talk
appears to exist between apoptosis and other cytotoxic mechanisms
ending in necrosis in which DNA laddering and caspase-8 activation
may not be strictly characterized as apoptotic events. Several
in vitro studies have shown that treatment of breast cancer
cells with various antiestrogens or aromatase inhibitors induces
cell death via unknown mechanisms (23–26).
Therefore, although DNA laddering and western blot analysis of
caspase-8 and -3 failed to demonstrate the activation of an
apoptotic mechanism as a means of cell death, a cytotoxic effect
mediated strictly by mechanisms resulting in necrosis may not be
accurate. As a result, subsequent studies may focus on determining
whether 4-OH-TAM-induced DNA adducts contribute to the observed
cytotoxic effect or whether autophagy or neroptosis are at play.
Furthermore, future aims may also include using endometrial cancer
cells to replicate studies conducted with breast cancer cells. When
analyzing TAM- and 4-OH-TAM-treated normal HMECs, apoptosis was
found in TAM-treated, but not 4-OH-TAM-treated HMECs (19). It would be of interest to
investigate whether similar results are noted with endometrial
cancer cells, thus elucidating the differences between TAM and
4-OH-TAM cytotoxic effects in the endometrium.
Due to the obvious decrease in cell survival
observed in both HEC-1B and HEC-1A cells exposed to 10 μM 4-OH-TAM
or 100 μM E2 for 1–3 days in comparison to the untreated
cells and the lack of apoptotic markers, we wanted to distinguish
whether the decline observed was due to a cytostatic (growth
inhibition) or cytotoxic (cell death) effect. After exposure to the
above concentrations of 4-OH-TAM and E2, a cytotoxic
effect was observed as determined by trypan blue dye exclusion
assay. Indeed, the number of living cells decreased to <25 and
0% for HEC-1B and HEC-1A cells treated with 4-OH-TAM and
E2, respectively. Therefore, the micromolar
concentrations of 4-OH-TAM and E2 induced a cytotoxic
effect resulting in extensive to complete cell death.
Overall, this study suggests that micromolar
concentrations of 4-OH-TAM induce a non-apoptotic cytotoxic effect
in the endometrium. However, subsequent studies are needed to
elucidate the underlying mechanism involved in the cytotoxic effect
of 4-OH-TAM and E2.
Acknowledgements
This study was supported by the MERCK-AAAS grant
awarded to the Biology and Chemistry Department, Sam Taylor
Fellowship and the Fleming Fund of Southwestern University awarded
to Dr Cuevas. The authors would like to thank Dr Maria Todd for her
editorial feedback, Taylor Vickers for his help with the figures,
Dr Cherryl Walker (MD Anderson, Smithville, TX) for providing the
HEC-1A and HEC-1B cell lines and Dr Romi Burks for her statistical
expertise.
References
|
1
|
Liu X, Pisha E, Tonetti DA, Yao D, Li Y,
Yao J, Burdette JE and Bolton JL: Antiestrogenic and DNA damaging
effects induced by tamoxifen and toremifene metabolites. Chem Res
Toxicol. 16:832–837. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Landel CC, Kushner PJ and Greene GL: The
interaction of human estrogen receptor with DNA is modulated by
receptor-associated proteins. Mol Endocrinol. 8:1407–1419.
1994.PubMed/NCBI
|
|
3
|
Stygar DN, Muravitskaya B, Eriksson H and
Sahlin L: Effects of SERM (selective estrogen receptor modulator)
treatment on growth and proliferation in the rat uterus. Reprod
Biol Endocrinol. 1:40–52. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Castro-Rivera E and Safe S:
17beta-Estradiol- and 4-hydroxy-tamoxifen-induced transactivation
in breast, endometrial and liver cancer cells is dependent on
ER-subtype, cell and promoter context. J Steroid Biochem Mol Biol.
84:23–31. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stackievicz R, Drucker L, Radnay J, Beyth
Y, Yarkoni S and Cohen I: Tamoxifen modulates apoptotic pathways in
primary endometrial cell cultures. Clin Cancer Res. 7:415–420.
2001.PubMed/NCBI
|
|
6
|
Goldenberg GJ and Froses EK: Drug and
hormone sensitivity of estrogen receptor positive and negative
human breast cancer cells in vitro. Cancer Res. 42:5147–5151.
1982.PubMed/NCBI
|
|
7
|
Taylor CM, Blanchard B and Zava DT:
Estrogen receptor mediated and cytotoxic effects of the
antiestrogens tamoxifen and 4-hydroxytamoxifen. Cancer Res.
44:1409–1414. 1984.PubMed/NCBI
|
|
8
|
Reddel RR, Murphy LC, Hall RE and
Sutherland RL: Differential sensitivity of human breast cancer cell
lines to the growth-inhibitory effects of tamoxifen. Cancer Res.
45:1525–1531. 1985.PubMed/NCBI
|
|
9
|
Lyman SD and Jordan VC: Metabolism of
tamoxifen and its uterotrophic activity. Biochem Pharmacol.
34:2787–2794. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pathak DN, Pongracz K and Bodell WJ:
Microsomal and peroxidase activation of 4-hydroxy-tamoxifen to form
DNA adducts: comparison with DNA adducts formed in Sprague-Dawley
rats treated with tamoxifen. Carcinogensis. 16:11–15. 1995.
View Article : Google Scholar
|
|
11
|
Kim SY, Suzuki N, Laxmi YR and Shibutani
S: Inefficient repair of tamoxifen-DNA adducts in rats and mice.
Drug Metab Dispos. 34:311–317. 2006. View Article : Google Scholar
|
|
12
|
Shibutani S, Ravindernath A, Suzuki N,
Terashima I, Sugarman SM, Grollman AP and Pearl ML: Identification
of tamoxifen-DNA adducts in the endometrium of women treated with
tamoxifen. Carcinogenesis. 21:1461–1467. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Castro-Rivera E and Safe S: Estrogen- and
antiestrogen-responsiveness of HEC-1A endometrial adenocarcinoma
cells in culture. J Steroid Biochem Mol Biol. 64:287–295. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Perry RR, Kang Y and Greaves B: Effects of
tamoxifen on growth and apoptosis of estrogen-dependent and
-independent human breast cancer cells. Ann Surg Oncol. 2:238–245.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Acconcia F, Barnes CJ and Kumar R:
Estrogen and tamoxifen induced cytoskeletal remodeling and
migration in endometrial cancer cells. Endocrinology.
147:1203–1212. 2006. View Article : Google Scholar
|
|
16
|
Carthew P, Edwards RE, Nolan BM, Tucker MJ
and Smith LL: Compartmentalized uterotrphic effects of tamoxifen,
toremifene and estradiol in the ovariectomized Wistar (Han) rat.
Toxicol Sci. 48:197–205. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dick GM, Rossow CF, Smirnov S, Horowitz B
and Sanders KM: Tamoxifen activates smooth muscle BK channels
through the regulatory β-1 subunit. J Biol Chem. 276:34594–34599.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thorneycroft IH Jr, Mishell DR, Stone SC,
Kharma KM and Nakamura RM: The relation of serum 17-OH progesterone
and estradiol-17-beta levels during the human menstrual cycle. Am J
Obstet Gynecol. 111:947–951. 1971.PubMed/NCBI
|
|
19
|
Dietze EC, Caldwell LE, Grupin SL, Mancini
M and Seewaldt VL: Tamoxifen but not 4-hydroxytamoxifen initiates
apoptosis in p53 (−) normal human mammary epithelial cells by
inducing mitochondrial depolarization. J Biol Chem. 276:5384–5394.
2001. View Article : Google Scholar
|
|
20
|
Vaux DL and Strasser A: The molecular
biology of apoptosis. Proc Natl Acad Sci USA. 93:2239–2244. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Catchpoole DR and Stewart BW:
Etoposide-induced cytotoxicity in two human T-cell leukemic lines:
delayed loss of membrane permability rather than DNA fragmentation
as an indicator of programmed cell death. Cancer Res. 53:4287–4296.
1993.PubMed/NCBI
|
|
22
|
Kanduc D, Mittelman A, Serpico R,
Sinigaglia E, Sinha AA, Natale C, Santacroce R, Di Corcia M,
Lucchese A, Dini L, Pani P, Santacroce S, Simon S, Bucci R and
Farberi E: Cell Death: Apoptosis versus necrosis (Review). Int J
Oncol. 21:165–170. 2002.PubMed/NCBI
|
|
23
|
Bouker KB, Sskaar TC, Fernandez DR,
O’Brien KA, Riggins RB, Cao D and Clarke R: Interferon regulatory
factor-1 mediates the proapoptotic but not cell cycle arrest
effects of the stroidal antiestrogen ICI 182,780 (Faslodex,
Fulvestrant). Cancer Res. 64:4030–4039. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
El Etreby MF, Liang Y, Wrenn RW and
Schoenlein PV: Additive effect of mifepristone and tamoxifen on
apoptotic pathways in MCF-7 human breast cancer cells. Breast
Cancer Res Treat. 51:149–168. 1998. View Article : Google Scholar
|
|
25
|
Thiantanawat A, Long BJ and Brodie AM:
Signaling pathways of apoptosis activated by aromatase inhibitors
and antiestrogens. Cancer Res. 63:8037–8050. 2003.PubMed/NCBI
|
|
26
|
Kallio A, Zheng A, Dahllund J, Heiskanen
KM and Härkönen P: Role of mitochondria in tamoxifen-induced rapid
cell death of MCF-7 breast cancer cells. Apoptosis. 10:1395–1410.
2005. View Article : Google Scholar : PubMed/NCBI
|