Introduction
Colorectal cancer (CRC) is a complex disease with
characteristics such as sustained proliferation, cell death evasion
and tissue invasion and metastasis, which make treatment difficult
(1,2). Cancer cell migration and invasion are
critical steps in the metastatic process and are regulated by
numerous cancer-secreted factors which modify the cancer
microenvironment by acting on stromal recruitment and extracellular
matrix (ECM) degradation (3).
Transforming growth factor βs (TGFβs) are 25-kDa
growth factors that play a unique and central role in homeostasis,
wound healing, fibrosis, angiogenesis, carcinogenesis and cell
differentiation (4,5). Each member of the TGFβ family is
encoded by different genes, although they act through the same
receptor-signaling cascade. They are stored in the ECM and attach
to latent TGFβ-binding proteins (6,7). This
attachment prevents the binding of the molecule to its receptor
(8).
During breast tumor progression, the loss of TGFβ
growth-inhibitory effects is frequently due to defects in c-myc and
p15 regulation by TGFβ (9).
However, other TGFβ responses are generally unrelated to growth
inhibition and favor tumor progression and metastasis (10–14).
Moreover, a study by Dai et al showed that p21 interacts
with Smad3/4 and the acetyl transferase p/CAF in order to regulate
Smad transcriptional activity, as well as gene transcription of
several other metastatic genes in breast cancer patients. These
results highlight the importance of p21/p/CAF-induced breast cancer
cell migration and invasion at the transcriptional level (15). In most CRC patients, TGFβ is
overexpressed and is likely associated with poor survival (16). A recent study showed that high p21
expression in pretreatment biopsies was associated with poor
prognosis in CRC patients treated with 5-fluorouracil (5-FU)-based
chemoradiotherapy (17).
Signaling from TGFβ through a transmembrane
serine-threonine kinase is an important Smad3/4 pathway, but plays
an ambiguous role in carcinogenesis (18–22).
The regulatory power of Smad3 as a transcriptional regulator is
augmented or modulated by interactions with ~50 co-transcription
factors (23). In addition, a study
by Ulloa et al reported a mechanism of transmodulation
between the STAT and SMAD signal-transduction pathways (24).
Previous studies have highlighted the important role
of TGFβ and Smads in various cancer types. However, the roles of
Smads downstream of TGFβ are still unknown in CRC and their
association with chemosensitivity has not been elucidated. The goal
of this research was to investigate how Smad3/4 are correlated with
chemotherapeutic drug sensitivity in human CRC and whether Smad3/4
and p21 are required to promote human CRC cell progression by TGFβ
signaling.
Materials and methods
Cell culture and reagents
The DLD-1, SNU-175, SNU-C4, Colo-320M, HT-29 and
HCT-15 human CRC cell lines were obtained from the Korean Cell Line
Bank (KCLB). DLD-1, SNU-175, SNU-C4, Colo-320M, HT-29 and HCT-15
were cultured in RPMI-1640 medium containing 10% fetal bovine serum
(FBS). All cells were cultured in a humidified incubator at 37°C
with 5% CO2. DLD-1 was made resistant to 5-FU by
incremental and continuous exposure to a formulation of 5-FU and
TGFβ1. Initially DLD-1 cells were treated with 10 μM 5-FU and 5
ng/ml TGFβ1 by limiting dilution. The resulting chemoresistant CRC
clone, named DLD1-5FU-C10, was able to grow in the presence of 75
μM of 5-FU in culture medium.
Cell viability inhibition by cytotoxic
agents
The CRC cell lines were seeded at 3×103
cells/well in 96-well white flat-bottomed plates. After incubation
for 24 h, CRC cells were treated with 5-FU or oxaliplatin at
various concentrations (0, 10, 100 and 1,000 or 0, 0.3, 0.6, 6, 12,
120 and 250 μM) in 10% FBS-supplemented RPMI-1640 for 72 h. The
toxicity of these treated cells was measured by adding 100 μl of
CellTiter-Glo® reagent (Promega, Madison, WI, USA) to
each well. Luminescence values in each well were determined using a
Spectra MAX plate reader (Molecular Devices, Sunnyvale, CA, USA).
Luminescence values from wells without cells (background) were
subtracted from the values of the wells with cells. Data were
analyzed with SigmaPlot software (Systat Software Inc., Chicago,
IL, USA) using Logistic 3 parameter analysis to determine the
half-maximal inhibitory concentration (IC50) of the
chemotherapeutic agents.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from the cells using TRIzol
reagent (Invitrogen, Grand Island, NY, USA) and reverse transcribed
into cDNA using the High Capacity RNA-to-cDNA kit (Applied
Biosystems, Grand Island, NY, USA) according to the manufacturer’s
instructions. The primer sequences were: interleukin 8 (IL8)
forward, GCAGAGGCCACCTGGATTG TGC and reverse,
TGGCATGTTGCAGGCTCCTCAGAA; (IL6) forward, CTCCCCTCCAGGAGCCCAGC and
reverse, GCAGGGAAGGCAGCAGGCAA; plasminogen activator (PLAU)
forward, GCCCTGGTTTGCGGCCATCT and reverse, CGCACACCTGCCCTCCTTGG;
matrix metalloproteinase 9 (MMP-9) forward, TGGACACGCACGACGTCT TCC
and reverse, TAGGTCACGTAGCCCACTTGGTCC; prostaglandin-endoperoxide
synthase 2 (PTGS2) forward, AGCTTTCACCAACGGGCTGGG and reverse,
AAGACCT CCTGCCCCACAGCAA; p21 forward, TGTCCGCGAGGA TGCGTGTTC and
reverse, GCAGCCCGCCATTAGCGCAT; GAPDH forward,
GCCTCAAGATCATCAGCAATGCCT and reverse, TGTGGTCATGAGTCCTTCCACGAT;
Smad3 forward, GGTCAAGAGCCTGGTCAAGA and reverse, TTG
AAGGCGAACTCACACAG; Smad4 forward, GACTGAGG TCTTTTCCGTTGG and
reverse, CTTCAAGCTCTGAGCC ATGC; STAT3 forward, GTGGGCGAGCGGTGTTCTG
and reverse, CAGAACACCGCTCGCCCAC; JAK1 forward, CAT
GGTGGAAGAGTTTGTGGAA and reverse, CAGCTGTTT GGCAACTTTGAATT. The
amplification conditions consisted of an initial denaturation at
95°C for 5 min, then 40 cycles of denaturation at 95°C for 30 sec,
annealing at 58°C for 30 sec and elongation at 72°C for 30 sec. A
1% agarose gel, containing Loading Star (DyneBionc, Gyeonggi,
Korea) for visualization, was run in Tris Borate-EDTA (TBE) buffer
for 20 min at 100 V, and the PCR products were analyzed using a Bio
Image Analyzer (Fisher Scientific, Seoul, Korea).
Small interfering RNA (siRNA)
DLD1-5FU-C10 cells were transfected with different
Smad3 and Smad4 siRNAs (AccuTarget™ Custom Designed siRNA; Bioneer,
Daejeon, Korea) and comprised the following targeting sequences:
Smad3 siRNA sense, 5′-GGAGAAAUGGUGCGAGAA Gtt-3′; Smad3 siRNA
antisense, 5′-CUUCUCGCACCAUUU CUCCtc-3′; Smad4 siRNA sense,
5′-GGUGGAGAGAGUGA AACAUtt-3′; and Smad4 siRNA antisense,
5′-AUGUUUCAC UCUCUCCACCtt-3′. For transient transfections,
105 cells were transfected with 100 nM siRNA using
Lipofectamine (Invitrogen).
Immunoblotting analysis
CRC cell lines and siRNA-transfected cells were
collected and lysed with Cell Lysis Buffer (Cell Signaling
Technology, Boston, MA, USA). Protein concentrations were
determined using a Pierce BCA protein assay kit (Thermal Scientific
Inc., Odessa, TX, USA). Equivalent amounts of protein from each
lysate were separated using SDS-PAGE and were transferred to
nitrocellulose membranes for immunoblotting. The membranes were
washed 3 times with Tris-buffered saline (TBS) containing 0.1%
Tween-20 (TBST). After blocking with TBST containing 5% nonfat milk
for 1 h, the membranes were incubated with the appropriate primary
antibody in TBST containing 3% skin milk at 4°C overnight. All of
the primary antibodies were diluted in an appropriate concentration
of 3% skim milk-containing TBST. After treatment with the primary
antibodies against Smad3, Smad4, p21 and IL-6 (all from Cell
Signaling Technology), IL-8 and PLAU (both from Abcam, Cambridge,
MA, USA), MMP-9, PTGS2, JAK1, STAT3 and β-actin (all from Cell
Signaling Technology), the membranes were washed 3 times with TBST
for 30 min, followed by goat anti-rabbit or anti-mouse
IgG-horseradish peroxidase-conjugated secondary antibody (diluted
at 1:4,000) for 2 h at room temperature and washed 3 times with
TBST for 1 h. The membranes were developed using the ECL western
blotting substrate (Promega) according to the manufacturer’s
instructions.
Cell migration assay
Cells were transfected with 3 siRNAs (negative
siRNA, Smad3 siRNA and Smad4 siRNA) and plated in 6-well plates at
106 cells/well. The 6-well plates ensured that images of
the wound could be automatically captured at the exact same
location by the Tsview 7 (Tucsen, Fuzhou, China). Cells were
scratched using a cell scraper (SPL, Pocheon, Korea) to generate
~250 μm-width wounds. After wounding, cells were washed 2 times
with PBS and 5-FU was added in the presence or the absence of 5
ng/ml of TGFβ. The plates were then placed into a Tsview 7 for 72
h. The data were analyzed by wound width or relative wound width
automatically measured by TSView 7 software (Tucsen).
Immunolocalization studies
CRC cell lines (105/ml) in 24-well plates
(Corning Inc., Corning, NY, USA) were washed 3 times with PBS,
fixed with 100% ethanol for 10 min on ice and then washed 3 times
with PBS. Cells were permeabilized with 0.025% Triton and blocked
for 1 h at room temperature with dilution buffer (Invitrogen).
Primary antibodies anti-Smad3, anti-Smad4 and anti-p21 (all from
Cell Signaling Technology) were then added to the dilution buffer
and incubated for 24 h at 4°C. The primary antibodies were removed
and the cells were washed 3 times for 3 min each with PBS. Next,
the cells were incubated with the appropriate secondary antibody
prepared in dilution buffer conjugated to FITC (1:500) for 4 h at
room temperature. Cells were washed again 3 times for 3 min each
with PBS and the cells were visualized using a Zeiss Observer Z1
AX10 (ZEISS, Oberkochen, Germany) fluorescence microscope.
Results
Anticancer drug sensitivity of human CRC
cell lines related to Smad3/4
The cytotoxic effects of 5-FU on 6 human CRC cell
lines (DLD-1, SNU-175, SNU-C4, Colo-320M, HCT-15, HT-29) were
examined using a luminescence assay (Fig. 1A). The IC50 values for
5-FU were 5, 6, 9, 40, 76 and 30 μM in the DLD-1, SNU-175, SNU-C4,
Colo-320M, HCT-15 and HT-29 cells, respectively. The cell viability
of the DLD-1, SNU-175 and SNU-C4 cell lines reflected high
sensitivity to 5-FU. The other cancer cell lines (Colo-320M,
HCT-15, HT-29) showed relatively low 5-FU sensitivity. The protein
expression levels of Smad3, Smad4 and p21 in the 6 human CRC cell
lines were determined by immunoblotting (Fig. 1B). Although all of the cancer cell
lines showed detectable levels of Smad3, Smad4 and p21, higher
levels of Smad3/4 and p21 protein were noted in the Colo-320M,
HCT-15 and HT-29 cells, which were the cell lines that showed
decreased 5-FU sensitivity (Fig.
1B). Since the DLD-1 cells showed low Smad3/4 and p21
expression and high 5-FU sensitivity, we selected DLD-1 cells to
proceed with the experiments and established resistance to 5-FU by
TGFβ treatment (DLD1-5FU-C10 cells).
Isolating chemoresistant human CRC cells
by TGFβ treatment
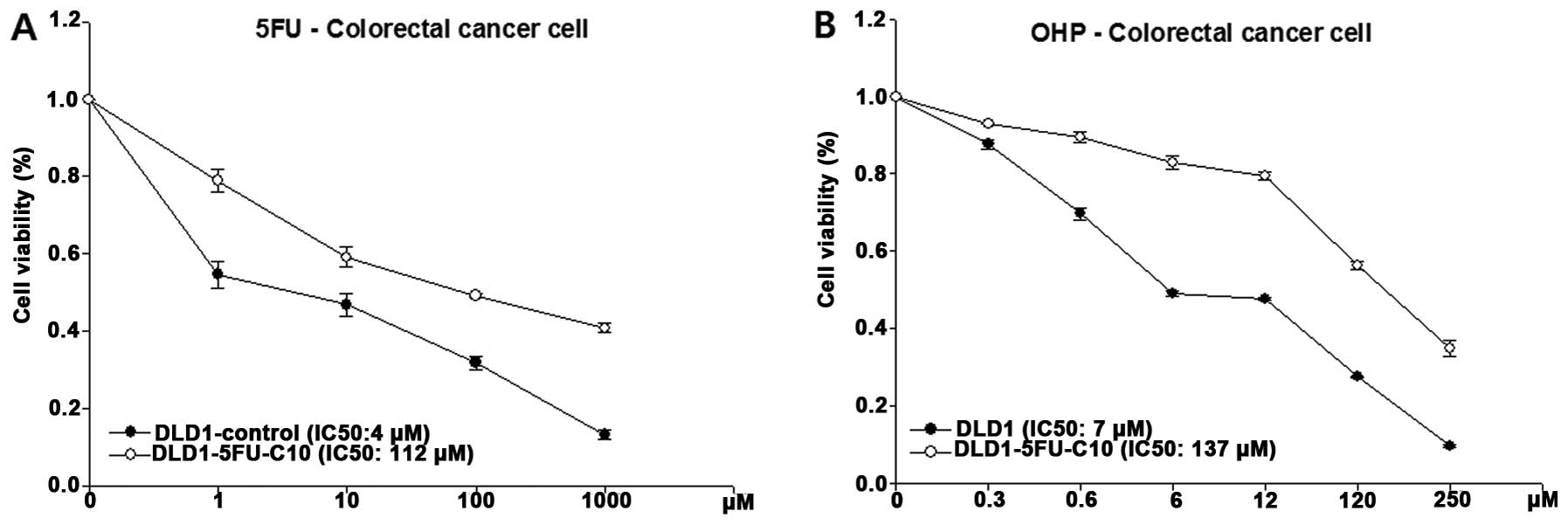
To confirm the cell viability to 5-FU in the
DLD1-5FU-C10 cells, we analyzed intrinsic sensitivity to 5-FU,
which resulted in a calculated IC50value of 112 μM for
5-FU (Fig. 2A). In addition,
DLD1-5FU-C10 cells showed decreased sensitivity to oxaliplatin, a
platinum-based antineoplastic agent, with a calculated
IC50 of 137 μM (Fig.
2B). Finally, we isolated DLD1-5FU-C10 cells that showed
IC50 values >10-fold higher than the DLD1
control.
Smad3/4 are related to drug sensitivity
and cell mobility via p21
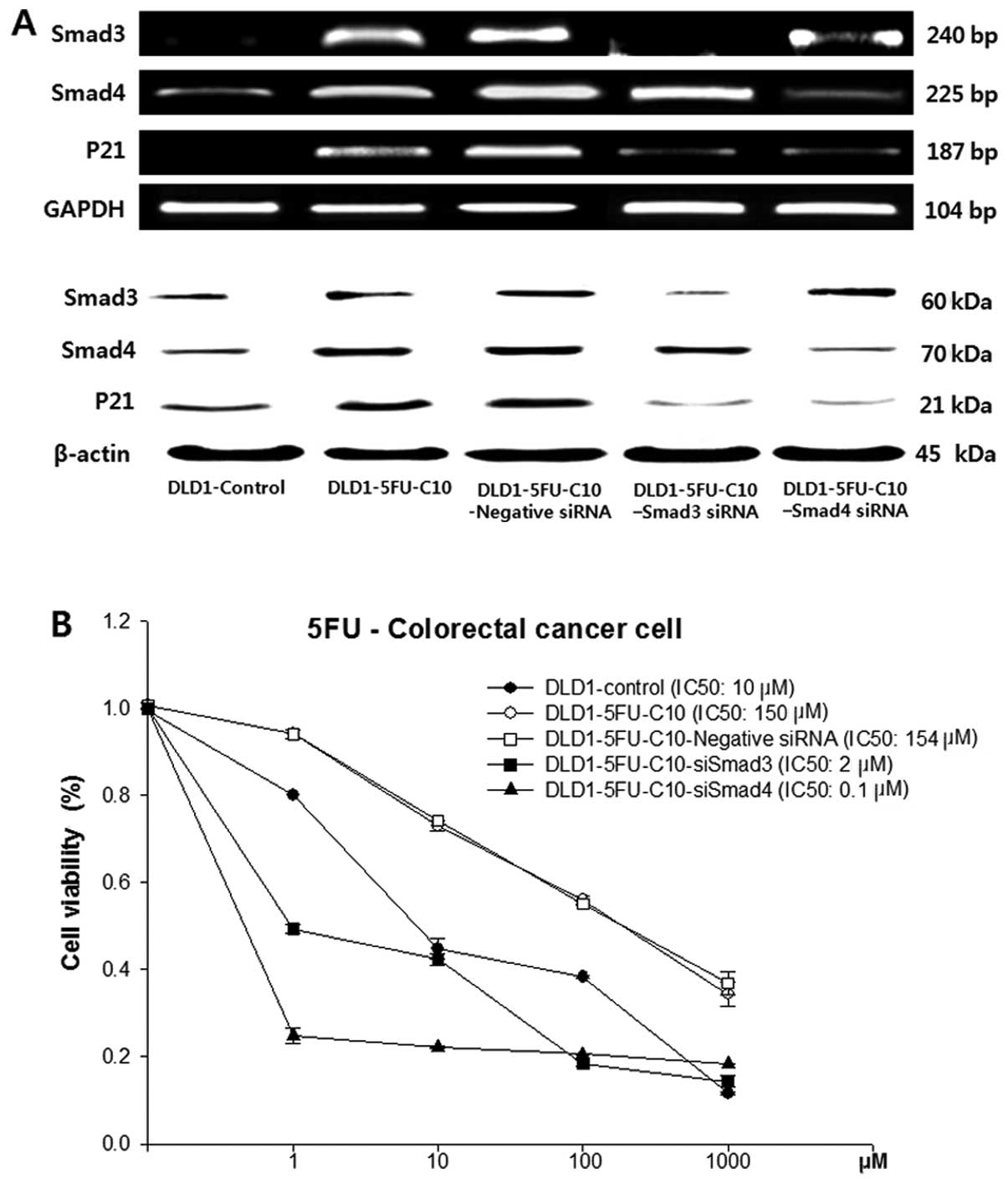
To evaluate further the relationship between Smad3/4
and drug sensitivity, Smad3/4 expression was knocked down by
Smad3/4 siRNAs in the DLD1-5FU-C10 cells. The knockdown was
confirmed by immunoblotting and RT-PCR (Fig. 3A).
Smad3/4 protein levels were decreased in the
DLD1-5FU-C10 cells treated with Smad3 and Smad4 siRNA when compared
with levels in the non-transfected DLD1-5FU-C10 cells or cells
treated with the negative siRNA. Smad3/4 siRNA caused slightly
lowered p21 expression when compared with that in the
non-transfected DLD1-5FU-C10 cells or the DLD1-5FU-C10 cells
treated with the negative siRNA. Therefore, our results indicated
that Smad3/4 down-regulation reduced p21 expression in the
DLD1-5FU-C10 cells. Knockdown of Smad3/4 expression in the
DLD1-5FU-C10 cells led to a decrease in cell viability and
IC50 values from 150 μM for the DLD1-5FU-C10 cells to
0.1 μM for the siSmad4 cells and 2 μM for the siSmad3 cells
(Fig. 3B).
We also investigated whether Smad3/4 are required
for cell migration in the DLD1-5FU-C10 cells using the
scratch/wound healing assay in the presence of 5-FU. Fig. 4 shows the migration of DLD1-5FU-C10
and DLD1-5FU-C10 cells with Smad3/4 knockdown (DLD1-5FU-C10-Smad3
siRNA, DLD1-5FU-C10-Smad4 siRNA). The rate of cell migration was
significantly higher in the DLD1-5FU-C10 cells than that in the
DLD1 control and Smad3/4 knockdown groups (Fig. 4). Wound closure was monitored by
measuring wound widths.
Chemoresistant human CRC cells induce
transcriptional activity of TGFβ downstream genes
Next, we performed signal pathway profiling
experiments in chemoresistant human CRC cells (DLD1-5FU-C10), using
transiently transfected Smad3/4 siRNA. We identified multiple
Smad3/4-dependent TGFβ target genes, among which we selected those
known to be associated with drug sensitivity. The shortlist
included 5 candidate target genes from our literature search
(15): IL6, IL8 (chemokine), PTGS2,
PLAU and MMP-9. The 5 TGFβ-induced downstream genes were detected
by immunoblotting (Fig. 5A) and
RT-PCR (Fig. 5B). As shown in
Fig. 5, DLD1-5FU-C10 cells showed
significantly higher mRNA expression of IL6, PLAU and PTGS2 than
did the DLD1 control cells. Furthermore, we analyzed the influence
of Smad3/4 knockdown on the DLD1-5FU-C10 cells, which showed a
recovery-signaling pathway to the DLD1 control.
 | Figure 5Effect of Smad3/4 on 5 TGFβ downstream
cytokines and the JAK/STAT pathway in low drug sensitivity human
colorectal cancer (CRC) cells. Smad3/4 affected 5 TGFβ downstream
cytokines and the JAK/STAT pathway in 5 groups of DLD-1 CRC cells
(DLD-1 control, DLD1-5FU-C10, DLD1-5FU-C10-Negative siRNA,
DLD1-5FU-C10-Smad3 siRNA, DLD1-5FU-C10-Smad4 siRNA). (A) Reverse
transcription-polymerase chain reaction (RT-PCR) of the expression
of 5 TGFβ downstream cytokines (IL8, IL6, PLAU, PTGS2 and MMP-9)
and the JAK1/STAT3 pathway (JAK1 and STAT3) in the 5 groups of DLD1
CRC cells. (B) Immunoblotting analysis of the expression of 3 TGFβ
downstream cytokines (IL6, PLAU and PTGS2) and the JAK1/STAT3
pathway in the 5 groups of DLD1 CRC cells. The same lysates were
also used to evaluate the expression of β-actin as a loading
control. IL, interleukin; PLAU, plasminogen activator; PTGS2,
prostaglandin endoperoxide synthase 2; MMP-9, matrix
metalloproteinase-9. |
Smad3/4 regulate STAT3 signaling in
chemoresistant human CRC cells
We showed that Smad3/4 induced the protein kinase
JAK1 and the transcription factor p-STAT3 in the chemoresistant
human CRC cells (DLD1-5FU-C10) via TGFβ (Fig. 5). Furthermore, Smad3/4 knockdown in
the DLD1-5FU-C10 cells decreased p-STAT3 signaling. Thus, we
hypothesized that the JAK1/STAT3 pathway could act downstream of
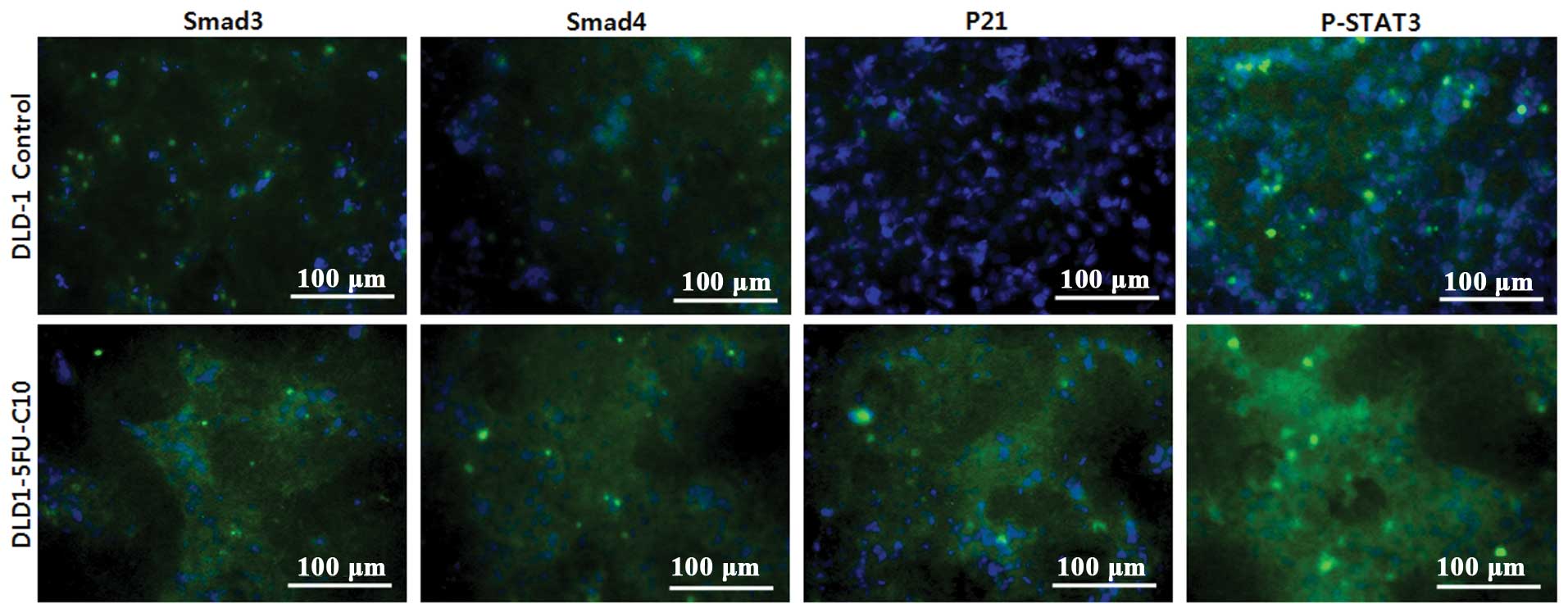
Smad3/4 to regulate anticancer drug sensitivity. We investigated
the localization of p21, Smad3/4 and p-STAT3 in the DLD1-5FU-C10
cells using immunocytochemistry (Fig.
6). The immunocytochemistry results showed that p-STAT3
signaling was increased in the chemoresistant human CRC cells.
Discussion
Smads are a class of proteins that function as
intracellular signaling effectors for TGFβ signaling (18). Smad2 and Smad3 are activated by
activin and TGFβ receptors, whereas Smad4 is activated by cytokine
receptors similar to the JAK/STAT signal transduction pathway
(18). In addition, a study by Dai
et al showed that Smad3/4 interact with p21 and are
associated with the poor prognosis of breast cancer patients
(15,25,26).
However, the effect of Smad3/4 on drug sensitivity in CRC has not
yet been established. In the present study, we investigated the
role of Smad3/4 in the drug sensitivity of CRC cells with
TGFβ-mediated resistance to 5-FU.
TGFβ is known to be a regulating factor in many
types of cancers. Following ligand-binding, the TGFβ receptor is
activated and phosphorylates 2 cognate Smads, Smad2 and Smad3,
which then bind to Smad4. The resulting complex then translocates
to the nucleus and regulates the expression of many genes by
binding to their promoters (23).
Previous studies have raised various questions regarding when the
TGFβ signaling pathway switches from tumor suppression to tumor
propagation (27).
Here, we found that the chemoresistant CRC cell line
DLD1-5FU-C10 was resistant to growth inhibition. In particular, we
found that high Smad3/4 expression was required for cell
proliferation and migration in the TGFβ-mediated chemoresistant CRC
cells (DLD1-5FU-C10). In agreement with these results, Smad3/4
knockdown using siRNA significantly decreased tumor propagation and
migration in the anticancer drug environment (Figs. 3 and 4). Collectively, these findings support
the notion of a chemotherapy-resistant pathway to Smad3/4 in CRC,
in accordance with the results of a previous study on breast cancer
(15). The breast cancer study
reported that Smad3/4 pro-migratory functions are mediated by p21
and that 5 major cytokines (IL6, IL8, PLAU, MMP-9 and PTGS2) were
induced (15). In the present
study, we showed that in the DLD1-5FU-C10 cells the protein levels
of 3 cytokines (IL6, PLAU and PTGS2) were increased by Smad3/4 and
p21 (Fig. 5A).
Studies have shown that IL6, secreted by lamina
propria T cells and macrophages, activated the JAK/STAT pathway
and promoted proliferation of tumor cells in a murine CRC model
(28,29). In addition, a study by Lee et
al showed that p-STAT3 signaling decreased anticancer drug
sensitivity in human glioma cells (30). Our results showed that Smad3/4 turns
on the JAK1/STAT3 pathway via IL6, since IL6 expression is
controlled by Smad3/4 (Fig. 5B).
Moreover, both cytoplasmic Smad3/4 and p21 were consequently
increased in p-STAT3 signaling (Fig.
6). Therefore, Smad3/4 have anti-apoptotic effects in the
anticancer drug environment, which modulate the gene transcription
of several TGFβ downstream cytokine genes (IL6, PLAU and PTGS2). In
particular, high expression of PLAU, which is important for
invasive growth, contributes to distinct aspects of cellular
transformation.
However, we do not know exactly how PLAU and PTGS2
function in conferring resistance to chemotherapy in CRC. It is
important to investigate whether Smad3/4-induced PLAU or PTGS2 is
correlated with any other pathways. Moreover, our experimental
results was limited to only one CRC cell line and anticancer drug,
and our resistant clone was made drug-resistant by TGFβ. Our
results suggest that Smad3/4 act as regulators of chemoresistance
in TGFβ mediated chemoresistant CRC cells and the association
between Smad3/4 and p21 is related to the JAK1/STAT3 pathway.
To summarize, we described the role of Smad3/4 in
chemoresistant CRC cells via p21. We showed that Smad3/4 interact
with p21 and regulate p-STAT3 signaling by IL6. In addition, we
identified Smad3/4 as a key factor in the JAK1/STAT3 pathway in
chemoresistant CRC progression in an anticancer environment.
Finally, these results highlight an important role for Smad3/4
signaling in anticancer drug sensitivity in CRC.
Acknowledgements
This study was supported by a grant (no.
02-2013-012) from the SNUBH Research Fund. The authors thank J.
Patrick Barron, Professor Emeritus, Tokyo Medical University and
Adjunct Professor, Seoul National University Bundang Hospital for
his editing of this manuscript.
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wels J, Kaplan RN, Rafii S and Lyden D:
Migratory neighbors and distant invaders: tumor-associated niche
cells. Genes Dev. 22:559–574. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Derynck R and Feng XH: TGF-beta receptor
signaling. Biochim Biophys Acta. 1333:F105–F150. 1997.PubMed/NCBI
|
|
5
|
Massague J: TGF-beta signal transduction.
Annu Rev Biochem. 67:753–791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Massague J: The transforming growth
factor-beta family. Annu Rev Cell Biol. 6:597–641. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roberts AB, Anzano MA, Lamb LC, Smith JM
and Sporn MB: New class of transforming growth factors potentiated
by epidermal growth factor: isolation from non-neoplastic tissues.
Proc Natl Acad Sci USA. 78:5339–5343. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sinha S, Nevett C, Shuttleworth CA and
Kielty CM: Cellular and extracellular biology of the latent
transforming growth factor-beta binding proteins. Matrix Biol.
17:529–545. 1998. View Article : Google Scholar
|
|
9
|
Chen CR, Kang Y and Massague J: Defective
repression of c-myc in breast cancer cells: A loss at the core of
the transforming growth factor beta growth arrest program. Proc
Natl Acad Sci USA. 98:992–999. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Derynck R, Akhurst RJ and Balmain A:
TGF-beta signaling in tumor suppression and cancer progression. Nat
Genet. 29:117–129. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Akhurst RJ and Derynck R: TGF-beta
signaling in cancer - a double-edged sword. Trends Cell Biol.
11:S44–S51. 2001.PubMed/NCBI
|
|
12
|
Wakefield LM and Roberts AB: TGF-beta
signaling: positive and negative effects on tumorigenesis. Curr
Opin Genet Dev. 12:22–29. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang B, Vu M, Booker T, et al: TGF-beta
switches from tumor suppressor to prometastatic factor in a model
of breast cancer progression. J Clin Invest. 112:1116–1124. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gong J, Ammanamanchi S, Ko TC and Brattain
MG: Transforming growth factor beta 1 increases the stability of
p21/WAF1/CIP1 protein and inhibits CDK2 kinase activity in human
colon carcinoma FET cells. Cancer Res. 63:3340–3346.
2003.PubMed/NCBI
|
|
15
|
Dai M, Al-Odaini AA, Arakelian A, Rabbani
SA, Ali S and Lebrun JJ: A novel function for p21Cip1 and
acetyltransferase p/CAF as critical transcriptional regulators of
TGFbeta-mediated breast cancer cell migration and invasion. Breast
Cancer Res. 14:R1272012. View
Article : Google Scholar
|
|
16
|
Tsushima H, Kawata S, Tamura S, et al:
High levels of transforming growth factor beta 1 in patients with
colorectal cancer: association with disease progression.
Gastroenterology. 110:375–382. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sim SH, Kang MH, Kim YJ, et al: P21 and
CD166 as predictive markers of poor response and outcome after
fluorouracil-based chemoradiotherapy for the patients with rectal
cancer. BMC Cancer. 14:2412014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Derynck R, Zhang Y and Feng XH: Smads:
transcriptional activators of TGF-beta responses. Cell. 95:737–740.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miyazono K, ten Dijke P and Heldin CH:
TGF-beta signaling by Smad proteins. Adv Immunol. 75:115–157. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Montgomery E, Goggins M, Zhou S, et al:
Nuclear localization of Dpc4 (Madh4, Smad4) in colorectal
carcinomas and relation to mismatch repair/transforming growth
factor-beta receptor defects. Am J Pathol. 158:537–542. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takenoshita S, Tani M, Mogi A, et al:
Mutation analysis of the Smad2 gene in human colon cancers using
genomic DNA and intron primers. Carcinogenesis. 19:803–807. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu J and Attisano L: Mutations in the
tumor suppressors Smad2 and Smad4 inactivate transforming growth
factor beta signaling by targeting Smads to the
ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 97:4820–4825.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Massague J, Seoane J and Wotton D: Smad
transcription factors. Genes Dev. 19:2783–2810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ulloa L, Doody J and Massague J:
Inhibition of transforming growth factor-beta/SMAD signalling by
the interferon-gamma/STAT pathway. Nature. 397:710–713. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xia W, Chen JS, Zhou X, et al:
Phosphorylation/cytoplasmic localization of p21Cip1/WAF1 is
associated with HER2/neu overexpression and provides a novel
combination predictor for poor prognosis in breast cancer patients.
Clin Cancer Res. 10:3815–3824. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee S and Helfman DM: Cytoplasmic p21Cip1
is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin
pathway. J Biol Chem. 279:1885–1891. 2004. View Article : Google Scholar
|
|
27
|
Lampropoulos P, Zizi-Sermpetzoglou A,
Rizos S, Kostakis A, Nikiteas N and Papavassiliou AG: TGF-beta
signalling in colon carcinogenesis. Cancer Lett. 314:1–7. 2012.
View Article : Google Scholar
|
|
28
|
Becker C, Fantini MC, Schramm C, et al:
TGF-beta suppresses tumor progression in colon cancer by inhibition
of IL-6 trans-signaling. Immunity. 21:491–501. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhong Z, Wen Z and Darnell JE Jr: Stat3: a
STAT family member activated by tyrosine phosphorylation in
response to epidermal growth factor and interleukin-6. Science.
264:95–98. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee ES, Ko KK, Joe YA, Kang SG and Hong
YK: Inhibition of STAT3 reverses drug resistance acquired in
temozolomide-resistant human glioma cells. Oncol Lett. 2:115–121.
2011.PubMed/NCBI
|