Introduction
Endometrial cancer (EC) is one of the most common
gynecological malignant tumors. In the United States, ~52,630 new
cases of EC will be diagnosed and 8,590 deaths are projected in
2014 (1). Although EC is
efficiently diagnosed and successfully treated, the treatment of
progesterone-resistant and aggressive EC is difficult, and is
associated with decreased survival rates (2,3). Women
with variants of endometrioid adenocarcinoma, high-grade histology,
deep myometrial invasion, a tumor that invades the uterine cervix,
or a tumor that spreads to a local or regional site have high risks
of metastasis and recurrence. Additionally, lower levels of
progesterone receptor B (PRB) are positively correlated with
progesterone resistance and poor prognosis. Therefore, reasonable
and effective treatment can significantly improve the survival of
patients with progesterone-resistant and aggressive EC. However,
the molecular mechanisms underlying EC are not yet fully
elucidated.
A novel cytokine system that promotes tumor
metastasis has been identified and is composed of the receptor
activator of nuclear factor-κB (RANK), its ligand RANKL and
osteoprotegerin (OPG). RANKL, a member of the tumor necrosis factor
(TNF) receptor superfamily, is expressed primarily in lymphoid
tissues and in osteoblastic cell lines (4). RANK is essential for the development
of lactating mammary glands (5,6) and
the formation of lymph nodes. Recent reports suggest that the
binding of RANKL to RANK triggers a host of intracellular reactions
through a TNF-related molecule signaling pathway, which promotes
the metastatic behavior of malignant epithelial cells (7–10). It
has been demonstrated that RANK/RANKL is associated with the
initiation of tumorigenesis, and the progression and invasion of
tumors in human mammary epithelial cells (11–13).
In addition, this system also promotes progestin-driven primary
mammary cancer development under the control of sex hormones
(11,12,14).
OPG, a decoy receptor, blocks the binding of RANKL to RANK (thus
preventing RANK activation), which is a secreted protein lacking
transmembrane domains.
It has been verified that medroxyprogesterone
acetate (MPA) can induce the expression of RANKL in the mammary
gland (12), which results in an
acceleration of tumorigenesis after multiparity or treatment with
progesterone and carcinogens (11).
Moreover, recent research has demonstrated that the RANK/RANKL
pathway is involved in the incidence of progesterone-driven breast
cancer. MPA has been identified as an efficacious and
well-tolerated treatment for EC for several decades. Intriguingly,
we found that RANK/RANKL was expressed in EC at levels similar to
those found in BC. The interaction of PRB/MPA and RANK/RANKL is
poorly understood in EC and warrants further elucidated.
In the present study, we performed the first
investigation of the relationship between RANK/RANKL expression in
EC tissues and clinicopathological features and revealed that
RANK/RANKL expression was markedly higher in malignant tumors than
that in normal endometrium. Then, we examined the effect of MPA on
cell behavior induced by RANKL in different EC cell lines (Ishikawa
and KLE), and we determined whether the PRB is associated with
progesterone resistance. We demonstrated that the RANK-RANKL system
can play a pivotal role in EC progression via the mitogen-activated
protein kinase (MAPK) pathway, which can be abrogated by the effect
of MPA-mediated PRB in Ishikawa cell. Given the promising results,
targeting of RANKL might serve as an appropriate therapy for EC,
particularly progesterone-resistant or aggressive EC.
Materials and methods
Tissue selection and patient
characteristics
Paraffin-embedded endometrial tissue samples were
collected from May 2011 to March 2013. Seventy tissue samples of EC
were obtained from patients, who underwent surgical staging and
tumor grading of the disease established in accordance with the
criteria of the Federation International of Gynecology and
Obstetrics (FIGO) staging system. Thirty normal endometrial tissues
were collected from patients who underwent hysterectomies due to
myoma, adenomyosis or other diseases. Twenty endometrial atypical
hyperplasia (EAH) samples were also selected from patients who
underwent hysteroscopy due to irregular vaginal bleeding. Twenty BC
samples were collected from patients following adenomammectomy.
None of the patients involved in the present study received
hormonotherapy or adjuvant radiation therapy before surgery.
Reagents and antibodies
Recombinant human RANK ligand was obtained from
PeproTech (Rocky Hill, NJ, USA). MPA was purchased from Sigma.
Rabbit anti-phospho-ERK1/2, -ERK1/2, -phospho-JNK, -JNK,
-phospho-p38, -p38, -RANK, -RANKL, -PRB, -NF-κB2, -H3, -Ki-67,
-PARP and -caspase-3 antibodies were purchased from Cell Signaling
Technology.
Immunohistochemical analysis
The expression levels of RANK and RANKL were
detected through the avidin-biotin complex/immunoperoxidase method.
Endometrial tissue samples were sliced into 4-μm-thick tissue
sections. The deparaffinized sections were then boiled in 1:50
ethylene diamine tetraacetic acid for antigen retrieval. The
specimens were incubated with 3% hydrogen peroxide. After blocking
with serum, the sections were incubated with primary antibodies
against RANK and RANKL. The antibody binding was detected using
reagents (Miao Tong, Shanghai, China). Two independent
investigators without knowledge of the clinical pathological
parameters evaluated the expression of RANK/RANKL. The assessment
method was as follows. Based on the staining intensity, the
sections were rated as negative (0), weak (1), medium (2), or
strong (3). In addition, combined with the extent of staining, the
sections were scored as 0 (0%), 1 (1–25%), 2 (26–50%), 3 (51–75%)
or 4 (76–100%). The sum of the intensity and extent of staining was
considered the final staining value of the paraffin sections. A
value of at least 4 was considered to indicate positive
expression.
Cell cultures
Human EC cell lines (Ishikawa and KLE) were
purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA). The cells were cultured in Dulbecco’s modified
Eagle’s medium-F12 (Gibco, Auckland, New Zealand) supplemented with
10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA), 100 μg/ml
streptomycin, and 100 U/ml penicillin (HyClone) at 37°C in a
humidified atmosphere of 5% CO2.
Vector construction and lentiviral
transduction
The human RANK gene (NM_001270949.1) was cloned into
pL-TO-IRES-LUC to construct a lentiviral vector overexpressing the
RANK gene (Nuo Bai, Shanghai, China). Ishikawa and KLE cells were
transduced with the viral supernatant of pL-TO-IRES-LUC (empty
vector, EV) and pL-TO-IRES-LUC-RANK, respectively, in the presence
of 8 μg/ml Polybrene. Stable cell lines were selected using BSD
(1.5 and 2 μg/ml). The clones were confirmed through quantitative
real-time polymerase chain reaction (qRT-PCR) and western blot
analysis.
Co-administration of MPA and RANKL
A total of 1×106 Ishikawa and
3×106 KLE cells were plated separately in 6-well plates.
The cells were treated with MPA, RANKL, MPA + RANKL, and an equal
volume of double-distilled water (control). After the treatments,
the proteins were extracted and detected by western blot
analysis.
Cell proliferation assay
The cell proliferation was assessed using a standard
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma, St. Louis, MO, USA) assay. The absorbance values were
calculated at 490 nm using a SpectraMax 190 microplate reader
(Bio-Rad Model 680).
Migration and invasion assays
A total of 1×105 cells were seeded into
the upper chamber of a 24-well chemotaxis chamber with
polycarbonate filters (8-μm pore) (Corning Incorporated, Glendale,
AZ, USA). The cells were treated with 1, 2 and 2.5 μg/ml RANKL for
4 h. The crystal violet-stained cells in five fields/well were
counted at ×200 magnification. The invasion of cells was performed
using Transwell chambers with 8-μm pore membranes coated with 40 μl
of Matrigel at a 1:6 dilution (BD Biosciences, San Jose CA, USA) on
the upper side. A total of 1×105 cells in 100 μl of
serum-free medium and 500 μl of complete medium were added to the
upper chamber and lower chamber, respectively. After incubation for
12, 24 and 48 h separately, the cells were fixed with 4%
paraformaldehyde and counted as described above.
Apoptosis analysis
MPA (10 μM), RANKL (1 μg/ml), MPA + RANKL, or an
equal volume of dimethyl sulfoxide (control) were added to the
cells, and the plates were incubated for 48 h. The cells were
digested, washed and resuspended in binding buffer. Annexin V-FITC
and PI were then added (BD Pharmingen, San Diego, CA, USA).
RNA extraction and real-time PCR
analysis
Total RNA was extracted with TRI reagent
(Invitrogen, Shanghai, China) and the quality of the total RNA was
assessed using a spectrophotometer (Pharmacia Biotech RNA/DNA
calculator). The RNA (500 ng) was reverse transcribed into cDNA
using a reverse transcription kit (Takara, Dalian, China). qRT-PCR
was performed with SYBR-Green Master Mix (Takara) on an ABI Prism
700 thermal cycler (Applied Biosystems, Foster City, CA, USA) to
detect the expression of various genes. The target gene expression
was calculated using the ΔΔCt method with β-actin as the
housekeeping gene. The following primers were used: β-actin,
5′-CAGCCATGTACGTTGCTATCCAGG-3′ and 5′-AGGTCCAGACGCAGGATGGCATG-3′;
PRB, 5′-TGCCCAGCATGTCGCCTTAG-3′ and 5′-CTGGCTTAGGGCTTGGCTTTC-3′;
RANK, 5′-AGCATTATGAGCATCTGGGACGG-3′ and
5′-CAGCAAGCATTTATCTTCTTCATTCC-3′.
Western blot analysis
For total protein extraction, the cells were
harvested and lysed with RIPA buffer containing protease and
phosphatase inhibitor cocktails. The nuclear protein was extracted
using NE-PER nuclear and cytoplasmic extraction reagents according
to the manufacturer’s protocol. We used the following antibodies:
ERK1/2, p-ERK1/2, JNK, p-JNK, p38, p-p38, NF-κB2 (p52, p100), H3,
caspase-3 and PARP. β-actin expression was used as the control.
Enzyme-linked immunosorbent assay
(ELISA)
OPG and RANKL serum concentrations were measured
using commercially available immunoenzymatic assay kits,
respectively, according to the manufacturer’s instructions (Ray
Biotech, Norcross, GA, USA; Abnova, Taipei, Taiwan). The ELISA kit
is an in vitro enzyme-linked immunosorbent assay for
quantitative measurement. A monoclonal antibody against the mouse
was employed as capture antibody and a biotinylated detection
polyclonal antibody from goat was used as the detection
antibody.
Nude mouse tumor xenograft assay
Twenty 6-week-old female nude BALB/c mice were
obtained from the Shanghai Life Science Institute (Slac Laboratory
Animal Co., Ltd., Shanghai, China). Animal research was carried out
in strict accordance with the recommendations in the Guideline for
the Care and Use of Laboratory Animals of China. The procedures
were approved by the Committee on the Ethics of Animal Experiments
of the Obstetrical and Gynecological Hospital affiliated Fudan
University [Permit Number: SYXK (hu) 2008-0064]. All efforts were
made to minimize animal suffering. In order to investigate the
interaction between MPA and RANKL in regards to tumor growth in
vivo, Ishikawa cells transfected with the lentiviral vector
carrying the human RANK gene were used. All mice were randomly
divided into four groups of five mice to receive 1×107
cells, and were treated with either vehicle (control), MPA (100
mg/kg body weight), RANKL (250 μg/kg) or MPA + RANKL, respectively.
The cells were injected subcutaneously into the groin of each
mouse. Subcutaneous tumor formation was monitored one week after
injection and was measured weekly using digital calipers. The
tumors were removed after 28 days. Tumor volume (mm3)
was calculated using the following formula: Tumor volume
(mm3) = (the longest diameter) × (the shortest
diameter)2 × 0.5.
Statistical analysis
The data were analyzed using the SPSS 17.0 software
(SPSS, Inc., Chicago, IL, USA). The statistical analyses were
performed using an unpaired Student’s t-test or one-way ANOVA. The
Chi-square test was employed to compare categorical data. All of
the experiments were performed in triplicate. P<0.05 was
considered statistically significant. All data are presented as
means with standard deviations (SD).
Results
RANK/RANKL is upregulated in EC and is
correlated with clinicopathological parameters
To determine the effect of RANK/RANKL expression on
the development and progression of EC, we performed
immunohistochemistry (IHC) on the endometrial tissues. The
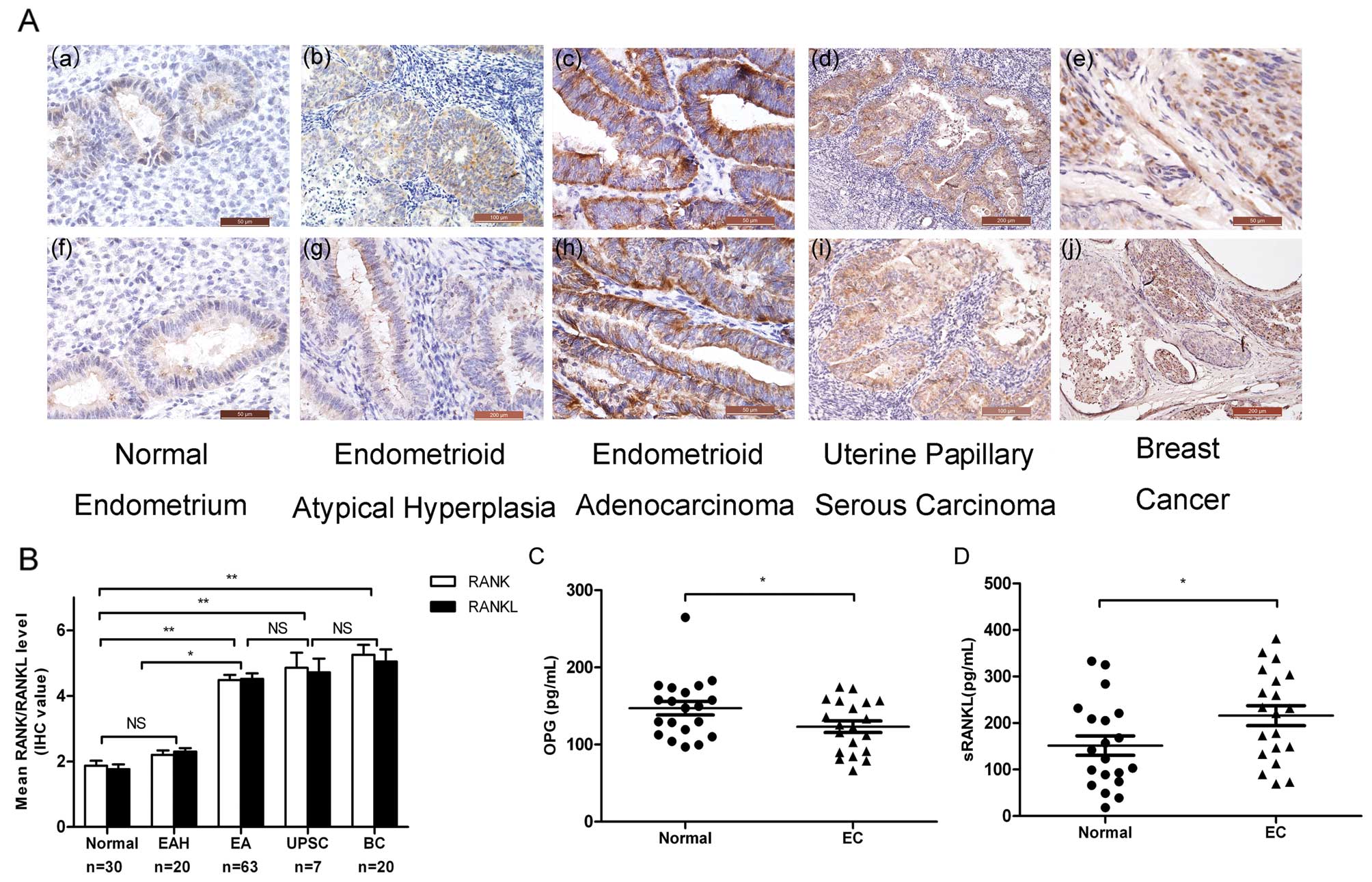
RANK/RANKL staining was predominantly localized to the cytoplasmic
membrane of the endometrial epithelial cells. Weak or no staining
was observed in the normal endometrium, and moderate to strong
RANK/RANKL staining was found in endometrial atypical hyperplasia
(EAH), endometrial adenocarcinoma (EA), uterine papillary serous
carcinoma (UPSC) and breast cancer (BC) (Fig. 1A). Briefly, most normal endometrium
samples were negative for RANK/RANKL (mean IHC value ≤2), and most
of the EAH samples exhibited weak staining (mean IHC value <3),
whereas the majority of the EC tissues presented positive
expression of RANK/RANKL (mean IHC value >4) (Fig. 1B). We explored the correlation
between RANK/RANKL expression and the clinicopathological
characteristics of EC. Higher RANK/RANKL expression levels were
observed in carcinomas with myometrial invasion (P=0.006; Table I), lymph node metastasis (P=0.045;
Table I) and lymphovascular space
involvement (P=0.025; Table I). We
did not, however, find any correlations between RANK/RANKL and
other clinicopathological characteristics, such as age, FIGO stage,
histological type, pathological grade or expression of estrogen
receptor (ER)/progesterone receptor (PR) (P>0.05; Table I). This result indicated that the
expression levels of RANK/RANKL play a pivotal role in the
risk-associated clinicopathological characteristics of EC.
RANK/RANKL overexpression increased the RANKL secretion in EC
patients, whereas the serum level of OPG was lower than that in the
healthy controls (Fig. 1C and
D).
 | Table ICorrelation between RANK/RANKL
expression and clinicopathological characteristics of the
endometrial cancer cases. |
Table I
Correlation between RANK/RANKL
expression and clinicopathological characteristics of the
endometrial cancer cases.
| | | RANK expression | RANKL expression |
|---|
| | |
|
|
|---|
| Parameters | Patients (n) | Patients (%) | Negative | Positive | P-value | Negative | Positive | P-value |
|---|
| Total | 70 | 100 | 13 | 57 | | 12 | 58 | |
| Age (years) | | | | | 0.452 | | | 0.088 |
| <50 | 16 | 22.9 | 4 | 12 | | 5 | 11 | |
| ≥50 | 54 | 77.1 | 9 | 45 | | 7 | 47 | |
| Grade
(endometrioid=63) |
| G1 or G2 | 57 | 90.5 | 8 | 49 | 0.219 | 9 | 48 | 0.955 |
| G3 | 6 | 9.5 | 2 | 4 | | 1 | 5 | |
| FIGO stage |
| I or II | 67 | 95.7 | 12 | 55 | 0.502 | 11 | 56 | 0.447 |
| III or IV | 3 | 4.3 | 1 | 2 | | 1 | 2 | |
| Histological
type |
| Endometrioid | 63 | 90 | 10 | 53 | 0.082 | 10 | 53 | 0.398 |
|
Non-endometrioid | 7 | 10 | 3 | 4 | | 2 | 5 | |
| Myometrial
invasion |
| <1/2 | 49 | 70 | 5 | 44 | 0.006a | 4 | 45 | 0.002a |
| ≥1/2 | 21 | 30 | 8 | 13 | | 8 | 13 | |
| Positive lymph
nodes |
| No | 42 | 60 | 11 | 31 | 0.045a | 11 | 31 | 0.014a |
| Yes | 28 | 40 | 2 | 26 | | 1 | 27 | |
| Lymphovascular
space involvement |
| No | 46 | 65.7 | 12 | 34 | 0.025a | 10 | 36 | 0.158 |
| Yes | 24 | 34.3 | 1 | 23 | | 2 | 22 | |
| ER expression |
| Negative | 15 | 21.4 | 4 | 11 | 0.363 | 1 | 14 | 0.225 |
| Positive | 55 | 78.6 | 9 | 46 | | 11 | 44 | |
| PR expression |
| Negative | 23 | 32.9 | 3 | 20 | 0.405 | 5 | 18 | 0.475 |
| Positive | 47 | 67.1 | 10 | 37 | | 7 | 40 | |
Determination of PRB expression in five
EC cell lines and verification of RANK overexpression
To confirm the endogenous PRB expression in EC cell
lines (Ishikawa, HEC-1B, AN3CA, RL95-2 and KLE), qRT-PCR and
western blot analysis were employed to determined the PRB mRNA and
protein level. The PRB protein was expressed at a low level in the
HEC-1B, AN3CA, RL95-2 cells, particularly in the KLE cells, and at
a relatively high level in the Ishikawa cells (Fig. 2A and B). Thus, we chose the Ishikawa
and KLE cells for further experiments. To explore the interaction
of RANK/RANKL with MPA/PRB, pL-TO-IRES-LUC-RANK was transfected
into the Ishikawa and KLE cells to construct stable cell lines that
overexpress the RANK gene (Fig. 2C and
D). Subsequently, qRT-PCR and western blot analysis were used
to verify the upregulation of RANK expression.
MPA influences the cell biological
behaviors of EC cells induced by RANKL in vitro
Accumulating evidence suggests the involvement of
RANK/RANKL in the regulation of cancer metastasis; thus, we aimed
to ascertain whether the upregulation of RANK influences the
behavior of Ishikawa and KLE cells. We tested the interaction
between MPA and RANKL in the cell viability of the EC cell lines.
MTT assays indicated that the MPA-treated cells had exceptionally
poor viability, whereas cell viability steadily increased after
RANKL treatment. The combination of MPA and RANKL decreased the
cell viability compared to RANKL treatment alone (data not shown).
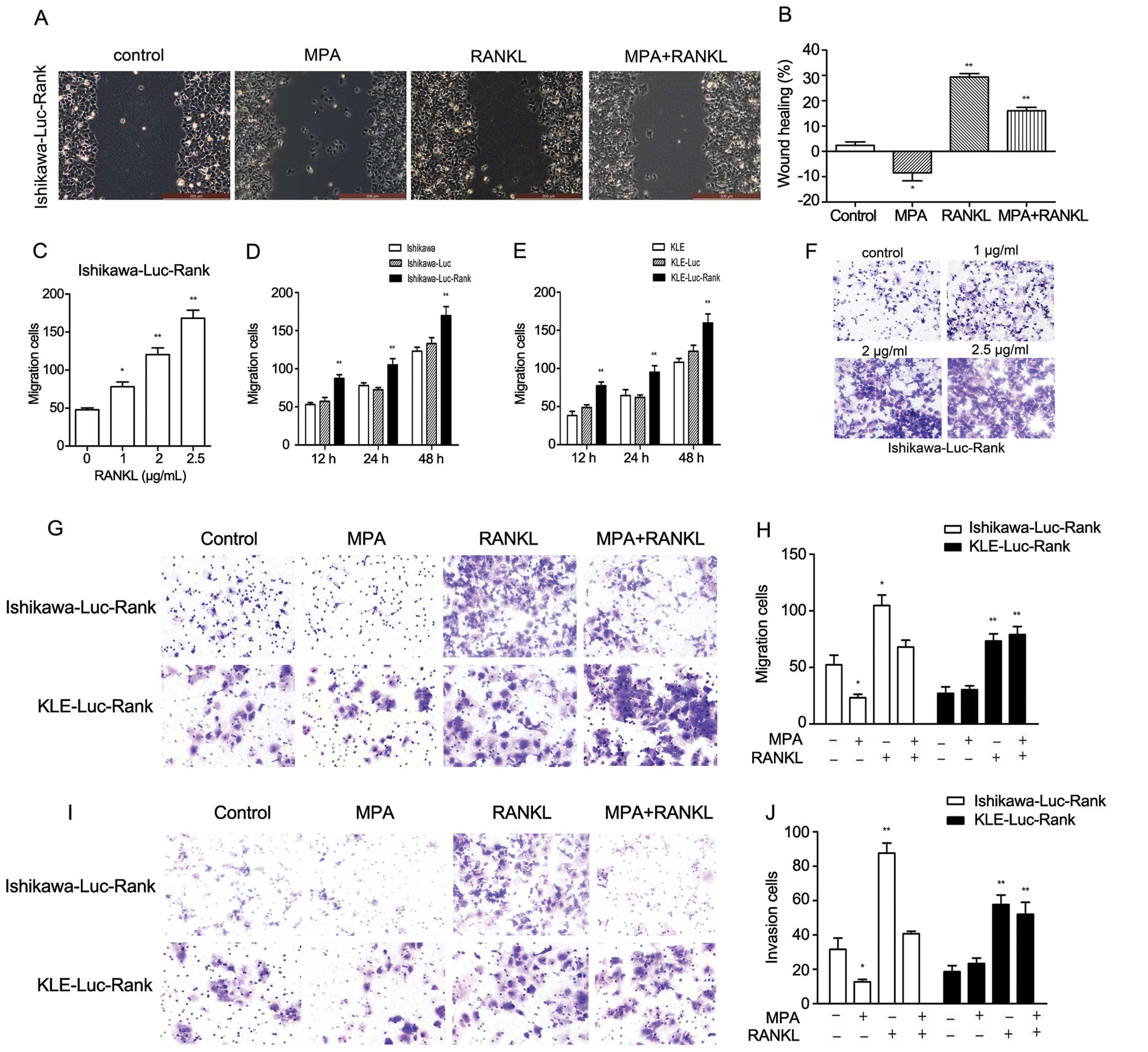
We also performed wound healing assays at 12 h after establishing
the wound. MPA-treated Ishikawa-Luc-Rank cells had a reduced wound
closure when compared with the control group, whereas RANKL-treated
Ishikawa-Luc-Rank cells had a greater extent of wound closure
(Fig. 3A and B).
To further investigate cell behaviors, we also
assayed the migratory and invasive capacities. There was a marked
increase in the migratory capacity of the Ishikawa-Luc-RANK and
KLE-Luc-Rank cells. Moreover, the effect of RANKL on cell migration
was dose- and time-dependent (Fig.
3C–F). The increase in cell migration in response to 1 μg/ml
RANKL was almost 40% compared to the control cells in the
Ishikawa-Luc-Rank cell line. To further explore the interaction
between MPA and RANKL in the migratory and invasive capacities,
Ishikawa-Luc-Rank cells and KLE-Luc-Rank cells were treated with
MPA, RANKL or MPA + RANKL. MPA obviously inhibited the migration
and invasion of the Ishikawa-Luc-Rank cells compared to the
untreated cells, whereas no effect was observed in the KLE-Luc-Rank
cells. The degree of invasion was markedly increased in response to
1 μg/ml RANKL in both cell lines. The combination of MPA and RANKL
also had no effect on inhibiting the migratory and invasive
capacities of KLE-Luc-Rank cells, whereas it reduced the migratory
and invasive numbers of Ishikawa-Luc-Rank cells (Fig. 3G–J). These findings collectively
indicated that the RANK-RANKL system may contribute to the
migration and invasion of EC cells and that MPA/PRB interfered with
the role of RANK/RANKL.
Effect of the RANK-RANKL pathway on the
apoptosis of EC cells
To elucidate the effects of MPA, RANKL and MPA +
RANKL on the apoptosis of these cells after a 48-h treatment, flow
cytometry and western blot analysis were performed. As demonstrated
in Fig. 4, MPA significantly
induced the apoptosis of the Ishikawa-Luc-Rank cells. However, the
administration of MPA and RANKL had no obvious promoting effects on
apoptosis compared to the MPA treatment. Thus, RANKL did not
promote the apoptosis-inducing effects of MPA in Ishikawa-Luc-Rank
cells. In addition, MPA almost had no effect on inducing apoptosis
of the KLE-Luc-Rank cells (Fig. 4A and
B).
To further elucidate the effects of RANK/RANKL on
apoptosis at the protein level, we detected the antibody levels of
caspase-3 and PARP. Consistent with the flow cytometry results, MPA
induced apoptosis through activation of caspase-3 and PARP, whereas
RANKL did not obviously promote apoptosis (Fig. 4C).
RANK-RANKL system activates the MAPK and
NF-κB signaling pathways
To further clarify the mechanisms responsible for
the RANK/RANKL regulation of tumor cell behavior, we noted that the
interaction between RANK and RANKL resulted in an obvious increase
in ERK1/2 and JNK phosphorylation levels in the Ishikawa-Luc-Rank
cells. A time-course analysis of the activation of the molecules in
the MAPK pathway after treatment with 1 μg/ml RANKL was performed
using the relevant antibodies. The results showed that exposure to
RANKL induced the phosphorylation of both ERK1/2 and JNK in the
Ishikawa-Luc-Rank and KLE-Luc-Rank cells, whereas there was no
significant change in the phosphorylation level of p38 (Fig. 5A). Meanwhile, we also demonstrated
the activation of the NF-κB pathway, since the activated form of
the NF-κB2 protein (p52) was detected in the cell nuclei (Fig. 5B). The effects of MPA, RANKL and MPA
+ RANKL on the activation of the MAPK pathway were further
examined. The Ishikawa-Luc-Rank cells demonstrated sensitivity to
MPA treatment. Conversely, the MPA-treated KLE-Luc-Rank cells did
not exhibit the inhibition of the phosphorylation levels of ERK1/2
and JNK. The combination of MPA and RANKL abrogated the activation
of ERK1/2 and JNK induced by RANK/RANKL in the Ishikawa-Luc-Rank
cells, whereas it had no effect on the KLE-Luc-Rank cells (Fig. 5C and D).
Oncogenic role of RANKL in a tumor
xenograft model
To further evaluate the effect of MPA, RANKL and MPA
+ RANKL on tumorigenesis in vivo, we measured tumor volumes
in xenografted mice over a 4-week period. These measurements
indicated that in the MPA− and MPA + RANKL-treated groups tumors
grew dramatically slower than those in the vehicle- or
RANKL-treated groups (Fig. 6A and
B). Four weeks after injection, the mice were sacrificed and
tumors were removed from the mice. The final mean body weights in
the vehicle and RANKL groups were lower than those in the other two
groups (Fig. 6C). These data
revealed that MPA efficiently inhibited endometrial tumor growth
in vivo, and MPA could also impede the oncogenic role of
RANKL. Tumor tissues were embedded in paraffin and stained with
hematoxylin and eosin (H&E). The expression levels of RANK,
RANKL and Ki-67 were determined by immunohistological assay
(Fig. 6D). In the MPA− and MPA +
RANKL-treated groups, lower expression of Ki-67 was consistent with
smaller tumor volumes.
Discussion
RANKL is regarded as a promising therapy strategy
for malignant tumors (6), and
agents that block RANKL to exacerbate antitumor effect have raised
considerable concern (15). Here,
we revealed that RANK/RANKL expression was significantly higher in
EC tissue than in normal endometrium and the expression of
RANK/RANKL was positively correlated with myometrial invasion,
lymph node metastasis, and lymphovascular space involvement,
suggesting that targeting of RANK/RANKL could be a therapy for
aggressive EC. Meanwhile, specific overexpression of RANK in
Ishikawa and KLE cells markedly increased the proliferation,
migration and invasion in vitro. Furthermore, MPA
efficiently inhibited the tumorigenicity in nude mice in
vivo. Therefore, we conclude that RANK/RANKL expression may
regulate EC-associated metastatic behavior.
The expression of PRB is essential to progesterone
action. Dai et al (16)
demonstrated that PR expression, particularly the expression of
PRB, appeared to be imperative for progesterone reaction, since the
inhibitory actions of progesterone on cell proliferation and
invasiveness may be due mainly to PRB activity. KLE has been
identified as a poorly differentiated EC cell line that lacks PRB
expression (17). Ai et al
(18) observed that MPA inhibited
the proliferation of Ishikawa cells, whereas it promoted the growth
of KLE cells due to their divergent PRB statuses. KLE is regarded
as an aggressive type 2 EC cell and progesterone-resistant EC cell
(hormone receptor-negative). Therefore, we used Ishikawa (high
levels of PRB) and KLE (low levels of PRB), which both have low
levels of RANK. Our data confirmed that the effect of MPA-mediated
PRB effectively inhibited the proliferation, migration and invasion
of the RANKL-induced Ishikawa-Luc-Rank cells in vitro,
whereas there was no change in the KLE-Luc-Rank cells. Thus, RANKL
may be an important therapeutic target for the treatment of
progesterone-resistant and aggressive EC.
The present study provides the initial evidence of
the expression and function of RANK/RANKL in EC. It was previously
proven that tumor cells that express RANK could activate the
RANK-RANKL pathway (19–21). Considering the extensive regulatory
role of RANK/RANKL, we constructed a cell-based model of high RANK
expression. In the present study, we demonstrated that the
stimulation of EC cells by RANKL activated specific downstream
pathways, including the ERK1/2, JNK and NF-κB pathways. In
addition, we explored the interaction between RANK/RANKL and
PRB/MPA treatment in terms of the biological behavior of EC cells
in vitro and in vivo.
Recent studies have highlighted that RANKL is
upregulated in a broad range of tumors and bone-related metastases,
mostly through the parathyroid hormone-related protein, which is
produced by hormones, cytokines and the tumor cells themselves
(22). The binding of RANKL to RANK
causes receptor trimerization and initiates a cascade of reactions
that facilitates the differentiation and maturity of tumor cells
(13). This action leads to the
enrollment of various molecules, including TNF receptor-associated
factor (TRAF)-6, which activates the MAPK pathway. The activation
of MAPK signaling cascades exerts a crucial role in transmitting
extracellular signals to regulate a variety of cellular responses
implicated in proliferation, differentiation, migration and
apoptosis (23–25). These studies provide new insights
into the critical cellular roles of RANKL in the development and
progression of cancer.
Activated MAP kinases which transform extracellular
stimuli into physiological responses phosphorylate molecules,
including cytoskeletal proteins and mitochondrial proteins, and
also regulate a wide range of transcription factors. NF-κB
proteins, which belong to the transcription factor superfamily, are
involved in preventing cells from undergoing apoptosis inducing by
DNA variation or cytokine treatment (26). Stimulation of the NF-κB pathway can
activate a series of downstream molecules and in turn lead to their
ubiquitination and degradation. These reactions can trigger the
translocation of NF-κB proteins and initiate the expression of
specific cellular genes. Accumulating evidence supports that NF-κB
proteins are associated with cellular proliferation and apoptosis,
which can be activated through the MAPK pathway (27,28).
We demonstrated that NF-κB signaling mediated the apoptosis in EC
cells induced by MPA, since the activated form of NF-κB2 protein
(p52) was detected in the cell nucleus.
OPG plays a role in blocking the interaction between
RANK and RANKL. We also demonstrated that the serum levels of OPG
in EC patients were lower than those in healthy controls, whereas
the levels of RANKL were higher. The reduction of serum levels of
OPG may be related to an enhancement of the interaction between
RANK and RANKL, which could activate the MAPK pathways to promote
tumor progression.
Based on the results discussed above, it is clearly
evident that RANK/RANKL is involved in the development and
progression of EC, providing new perspectives in the analysis of
the biological mechanisms of EC. MPA/PRB administration plays a
vitally important role in the inhibition of the proliferation,
migration, and invasion of EC cells. Drug targeting of RANKL may be
an appealing candidate for progesterone-resistant and aggressive
EC. Further studies are warranted to investigate the applications
and implications in future clinical work and research.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (nos. 81172477 and 81072140),
the Project of the Science and Technology Commission of Shanghai
Municipality (nos. 11ZR1440800 and 13JC1401303), and the Project of
Outstanding Subject Leaders of the Shanghai Health System (no.
XBR2013097).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Di Cristofano A and Ellenson LH:
Endometrial carcinima. Annu Rev Pathol. 2:57–85. 2007. View Article : Google Scholar
|
|
3
|
Salvesen HB, Haldorsen IS and Trovik J:
Markers for individualised therapy in endometrial carcinoma. Lancet
Oncol. 13:e353–e361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kong YY, Yoshida H, Sarosi I, Tan HL,
Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G,
Itie A, et al: OPGL is a key regulator of osteoclastogenesis,
lymphocyte development and lymph-node organogenesis. Nature.
397:315–323. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fata JE, Kong YY, Li J, Sasaki T,
Irie-Sasaki J, Moorehead RA, Elliott R, Scully S, Voura EB, Lacey
DL, et al: The osteoclast differentiation factor
osteoprotegerin-ligand is essential for mammary gland development.
Cell. 103:41–50. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beleut M, Rajaram RD, Caikovski M, Ayyanan
A, Germano D, Choi Y, Schneider P and Brisken C: Two distinct
mechanisms underlie progesterone-induced proliferation in the
mammary gland. Proc Natl Acad Sci USA. 107:2989–2994. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Armstrong AP, Tometsko ME, Glaccum M,
Sutherland CL, Cosman D and Dougall WC: A RANK/TRAF6-dependent
signal transduction pathway is essential for osteoclast
cytoskeletal organization and resorptive function. J Biol Chem.
277:44347–44356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Darnay BG, Haridas V, Ni J, Moore PA and
Aggarwal BB: Characterization of the intracellular domain of
receptor activator of NF-kappaB (RANK). Interaction with tumor
necrosis factor receptor-associated factors and activation of
NF-kappaB and c-Jun N-terminal kinase. J Biol Chem.
273:20551–20555. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mori K, Le Goff B, Berreur M, Riet A,
Moreau A, Blanchard F, Chevalier C, Guisle-Marsollier I, Léger J,
Guicheux J, et al: Human osteosarcoma cells express functional
receptor activator of nuclear factor-kappa B. J Pathol.
211:555–562. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang L, Teng Y, Zhang Y, Liu J, Xu L, Qu
J, Hou K, Yang X, Liu Y and Qu X: C-Src-mediated RANKL-induced
breast cancer cell migration by activation of the ERK and Akt
pathway. Oncol Lett. 3:395–400. 2012.PubMed/NCBI
|
|
11
|
Gonzalez-Suarez E, Jacob AP, Jones J,
Miller R, Roudier-Meyer MP, Erwert R, Pinkas J, Branstetter D and
Dougall WC: RANK ligand mediates progestin-induced mammary
epithelial proliferation and carcinogenesis. Nature. 468:103–107.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schramek D, Leibbrandt A, Sigl V, Kenner
L, Pospisilik JA, Lee HJ, Hanada R, Joshi PA, Aliprantis A,
Glimcher L, et al: Osteoclast differentiation factor RANKL controls
development of progestin-driven mammary cancer. Nature. 468:98–102.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Palafox M, Ferrer I, Pellegrini P, Vila S,
Hernandez-Ortega S, Urruticoechea A, Climent F, Soler MT, Muñoz P,
Viñals F, et al: RANK induces epithelial-mesenchymal transition and
stemness in human mammary epithelial cells and promotes
tumorigenesis and metastasis. Cancer Res. 72:2879–2888. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schramek D, Sigl V and Penninger JM: RANKL
and RANK in sex hormone-induced breast cancer and breast cancer
metastasis. Trends Endocrinol Metab. 22:188–194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Santini D, Schiavon G, Vincenzi B, Gaeta
L, Pantano F, Russo A, Ortega C, Porta C, Galluzzo S, Armento G, et
al: Receptor activator of NF-kB (RANK) expression in primary tumors
associates with bone metastasis occurrence in breast cancer
patients. PLoS One. 6:e192342011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dai D, Wolf DM, Litman ES, White MJ and
Leslie KK: Progesterone inhibits human endometrial cancer cell
growth and invasiveness: down-regulation of cellular adhesion
molecules through progesterone B receptors. Cancer Res. 62:881–886.
2002.PubMed/NCBI
|
|
17
|
Leslie KK, Kumar NS, Richer J, Owen G,
Takimoto G, Horwitz KB and Lange C: Differential expression of the
A and B isoforms of progesterone receptor in human endometrial
cancer cells. Only progesterone receptor B is induced by estrogen
and associated with strong transcriptional activation. Ann NY Acad
Sci. 828:17–26. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ai ZH, Wang J, Wang YD, Lu LH, Tong JQ and
Teng YC: Overexpressed epidermal growth factor receptor
(EGFR)-induced progestin insensitivity in human endometrial
carcinoma cells by the EGFR/mitogen-activated protein kinase
signaling pathway. Cancer. 116:3603–3613. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jones DH, Nakashima T, Sanchez OH,
Kozieradzki I, Komarova SV, Sarosi I, Morony S, Rubin E, Sarao R,
Hojilla CV, et al: Regulation of cancer cell migration and bone
metastasis by RANKL. Nature. 440:692–696. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Armstrong AP, Miller RE, Jones JC, Zhang
J, Keller ET and Dougall WC: RANKL acts directly on RANK-expressing
prostate tumor cells and mediates migration and expression of tumor
metastasis genes. Prostate. 68:92–104. 2008. View Article : Google Scholar
|
|
21
|
Mori K, Le Goff B, Charrier C, Battaglia
S, Heymann D and Rédini F: DU145 human prostate cancer cells
express functional receptor activator of NFkappaB: new insights in
the prostate cancer bone metastasis process. Bone. 40:981–990.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wittrant Y, Théoleyre S, Chipoy C,
Padrines M, Blanchard F, Heymann D and Rédini F: RANKL/RANK/OPG:
new therapeutic targets in bone tumours and associated osteolysis.
Biochim Biophys Acta. 1704:49–57. 2004.PubMed/NCBI
|
|
23
|
Nebreda AR and Porras A: p38 MAP kinases:
beyond the stress response. Trends Biochem Sci. 25:257–260. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schieven GL: The biology of p38 kinase: a
central role in inflammation. Curr Top Med Chem. 5:921–928. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Manning AM and Davis RJ: Targeting JNK for
therapeutic benefit: from junk togold? Nat Rev Drug Discov.
2:554–565. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yamamoto Y and Gaynor RB: Therapeutic
potential of inhibition of the NF-kappaB pathway in the treatment
of inflammation and cancer. J Clin Invest. 107:135–142. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barkett M and Gilmore TD: Control of
apoptosis by Rel/NFkappaB transcription factors. Oncogene.
18:6910–6924. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hashimoto I, Koizumi K, Tatematsu M,
Minami T, Cho S, Takeno N, Nakashima A, Sakurai H, Saito S, Tsukada
K and Saiki I: Blocking on the CXCR4/mTOR signaling pathway induces
the anti-metastatic properties and autophagic cell death in
peritoneal disseminated gastric cancer cells. Eur J Cancer.
44:1022–1029. 2008. View Article : Google Scholar : PubMed/NCBI
|