Introduction
Oral squamous cell carcinoma is the most common head
and neck cancer. It accounts for more than 90% of all head and neck
cancers and has a poor prognosis (1). Tongue squamous cell carcinoma (TSCC)
is the most common oral squamous cell carcinoma. It is associated
with poorer survival and a lower rate of local tumor control than
other sites of head and neck cancer, with a 5-year survival rate of
50% (2). Chemotherapy has been
widely accepted as one of the three major therapies (surgery,
chemotherapy and radiotherapy) treating advanced squamous cell
carcinoma in the head and neck region (3). Cisplatin, a commonly used
chemotherapeutic agent, is effective as a single agent or in
combination with other drugs for the treatment of TSCC (4). Treatment with cisplatin-based
chemotherapy has been found to improve the prognosis of patients
with TSCC (5). However, one of the
most important clinical issues of cisplatin-based TSCC chemotherapy
is the intrinsic/acquired chemoresistance to cisplatin (6).
Traditionally, the targets and modulators of
chemotherapy most in focus are DNA, mRNA and proteins (3). Recently, it has been found that
microRNAs (miRNAs) are implicated in oncogenic cell processes
including chemoresistance (7).
miRNAs are small non-coding RNA molecules of 19–25 nucleotides in
length. They regulate target gene expression post-transcriptionally
by incomplete base pairing with their target mRNAs (8). They function through RNA-induced
silencing complexes, targeting them to mRNAs where they either
repress translation or direct destructive cleavage (9). A recent study has shown that the
increased expression of miR-23a promotes cisplatin chemoresistance
in TSCC cells (3).
Wist, also known as Twist1, belongs to the basic
helix-loop-helix transcription factor family. High expression of
Twist has been detected in several types of cancers and has been
associated with cancer progression and chemoresistance (10). A recent study has shown that
overexpression of Twist is associated with poor prognosis of TSCC
patients (11), which suggests that
Twist could be a potential therapeutic target for TSCC.
Our pilot study suggested that miR-23a regulates the
expression of Twist in TSCC cells. In the present study, we
explored the interaction between miR-23a and Twist in TSCC cells,
and assessed its impact on TSCC chemoresistance to cisplatin.
Materials and methods
Cell lines, plasmids and reagents
Human TSCC cell lines SCC-4 and Tca8113 were
purchased from the American Tissue Culture Collection (ATCC;
Manassas, VA, USA) and the Wuhan Boster Biological Engineering Inc.
(Wuhan, China), respectively. Twist (sc-38604-V) shRNA lentiviral
particles, control shRNA lentiviral particles-A (sc-108080) and
anti-Twist (sc-81417) and anti-β-actin (ACTBD11B7) (sc-81178)
antibodies were purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). SuperFect transfection reagent was purchased from
Qiagen (Valencia, CA, USA). The non-radioactive c-Jun N-terminal
kinase (JNK) assay kit (#8794) was purchased from New England
Biolabs (Beverly, MA, USA). Human Twist cDNA was subcloned into the
pcDNA 3.1 expression vector (12).
The pMIRH23aPA-1 lentiviral vector expressing miR-23a, the
pMIRZIP-23a lentiviral vector expressing shRNA against miR-23a, and
the lentiviral packaging system were purchased from System
Biosciences (Mountain View, CA, USA). The Annexin V-EGFP apoptosis
detection kit was purchased from BioVision (Mountain View, CA,
USA). Puromycin, cisplatin, selective JNK inhibitor SP600125 and
all chemicals of reagent grade were purchased from Sigma (St.
Louis, MO, USA).
Transfection and lentiviral
transduction
The Twist expression vector was transfected into
cells using SuperFect transfection reagent according to the
manufacturer’s instructions. Pools of stable transductants were
generated via selection with G418 (600 μg/ml) by the manufacturer’s
protocol. Lentiviral transduction of Twist-shRNA was performed in
SCC-4 and Tca8113 cells. Pools of stable transductants were
generated via selection with puromycin (4 μg/ml) according to the
manufacturer’s protocol (Santa Cruz Biotechnology). SCC-4 and
Tca8113 cells were also transduced with the lentivirus expressing
miR-23a (pMIRH23aPA-1) sequences, shRNAs against miR-23a
(pMIRZIP-23a), or control scrambled hairpin vector sequences under
the control of constitutive H1 promoter (Systems Biosciences).
Lentiviral supernatants were used to infect the cells with the
addition of Polybrene at 8 μg/ml for 8 h. Cells were harvested 48 h
after the lentiviral transduction.
Real-time quantitative reverse
transcription PCR
Total RNA was prepared from cells using TRIzol
reagent followed by purification with TURBO DNA-free system
(Ambion, Austin, TX, USA). The cDNAs were synthesized using
SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA,
USA). Real-time quantitative PCR was performed using SYBR-Green PCR
Master Mix in a 7300 Real-Time PCR system (both from Applied
Biosystems, Foster City, CA, USA). TaqMan microRNA assays (Applied
Biosystems) that include RT primers and TaqMan probes were used to
quantify the expression of miRNA-23a; RNU66 (PN 4373382) was used
as an internal control (Applied Biosystems) for determining the
expression level of miR-23a. For measurement of the Twist mRNA
level, the following primers were used: for Twist, 5′-AC
GAGCTGGACTCCAAGATG-3′ (forward) and 5′-CACGCC CTGTTTCTTTGAAT-3′
(reverse); for glyceraldehyde-3-phosphate dehydrogenase (GAPDH),
5′-GACTCATGACCACAGT CCATGC-3′ (forward) and 5′-AGAGGCAGGGATGATGTT
CTG-3′ (reverse). The results were normalized against that of the
GAPDH gene in the same sample. Each experiment was repeated for
three times in duplicates.
Western blot analysis
Briefly, cells were dissolved in 250 μl of 2X SDS
loading buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 25% glycerol,
0.01% bromphenol blue, 5% 2-mercaptoethanol), and incubated at 95°C
for 10 min. Equal amount of proteins for each sample were separated
by 10% SDS-polyacrylamide gel and blotted onto a polyvinylidene
difluoride microporous membrane (Millipore, Billerica, MA, USA).
Membranes were incubated for 1 h with a 1/500 dilution of primary
antibody, and then washed and revealed using secondary antibodies
conjugated with horseradish peroxidase (1/5,000; 1 h). Peroxidase
was revealed with a GE Healthcare ECL kit (Shanghai, China).
JNK activity assay
The JNK activity was measured with a non-radioactive
assay kit as per the manufacturer’s protocol (New England Biolabs)
(13). Briefly, JNK was
precipitated from the cell lysate by c-Jun fusion protein bound to
glutathione sepharose beads. c-Jun contains a high affinity-binding
site for JNK, N-terminal to the two phosphorylation sites, Ser63
and Ser73. After selectively pulling down JNK using c-jun fusion
protein beads, the beads were extensively washed and the kinase
reaction was carried out in the presence of cold ATP in a final
volume of 25 μl. The reaction was stopped with 25 μl of 2X SDS
sample buffer and loaded onto a 10% polyacrylamide gel. Protein was
transferred to nitrocellulose by electroblotting, and c-jun
phosphorylation was selectively measured using the phospho-c-Jun
antibody.
Cisplatin
chemosensitivity/chemoresistance assay
Cells were plated in duplicates in 96-well plates at
a density of 5,000 cells. After 24 h of incubation, the medium was
replaced by fresh medium with or without various concentrations of
cisplatin (Sigma). Then cell viability was assayed 48 h later using
a modified MTT assay as previously described (14). The half maximal inhibitory
concentration (IC50) was defined as the concentration
resulting in a 50% reduction in growth compared to control
cells.
Apoptosis analysis
Cells were seeded at 5×105/well in 6-well
plates. After cells attached to the plates (~2 h after seeding),
cells were treated with 20 μM of cisplatin for 30 h. Cell apoptosis
was analyzed with an Annexin V-EGFP apoptosis detection kit coupled
with flow cytometric analysis according to the manufacturer’s
instructions (BioVision).
Statistical analysis
Statistical analyses were performed with SPSS for
Windows 10.0 (IBM, Chicago, IL, USA). All data values are expressed
as means ± SD. Comparisons of means among multiple groups were
performed with one-way ANOVA followed by post-hoc pairwise
comparisons using Tukey’s test. A two-tailed p<0.05 was
considered to indicate a statistically significant result in the
present study.
Results
Overexpression and knockdown of miR-23a
and Twist in human TSCC cells
To investigate the functional interaction between
miR-23a and Twist in TSCC cells, we overexpressed miR-23a and Twist
in SCC-4 and Tca8113 human TSCC cells, and on the other hand
transduced the cells with lentiviral shRNAs to knock down miR-23a
and Twist, respectively. Real-time reverse transcription PCR showed
that compared with the controls, miR-23a was overexpressed by
~3.5-fold and knocked down ~70% in SCC-4 and Tca8113 cells,
respectively; the Twist mRNA was overexpressed by ~4-fold and
knocked down ~75% in SCC-4 and Tca8113 cells, respectively
(Fig. 1). Twist expression at both
the mRNA (Fig. 1) and protein
levels (Fig. 2) in the TSCC cells
increased (by ~3-fold at the mRNA and 2.6-fold at the protein
level) and decreased (by ~53% at the mRNA and 50% at the protein
level) in parallel with miR-23a overexpression and knockdown,
respectively. In contrast, overexpression and knockdown of Twist
had no significant effect on the expression of miR-23a (Fig. 1). Our pilot study suggested that
miR-23a would regulate Twist expression in TSCC cells by a
JNK-dependent mechanism (data not shown). Therefore, we included a
selective JNK inhibitor SP600125 (10 μM) in all experiments in the
present study (15). As shown in
Fig. 1, the JNK inhibitor had no
significant effect on the constitutive expression level of miR-23a.
However, it abolished miR-23a-induced Twist expression in the TSCC
cells (Figs. 1 and 2).
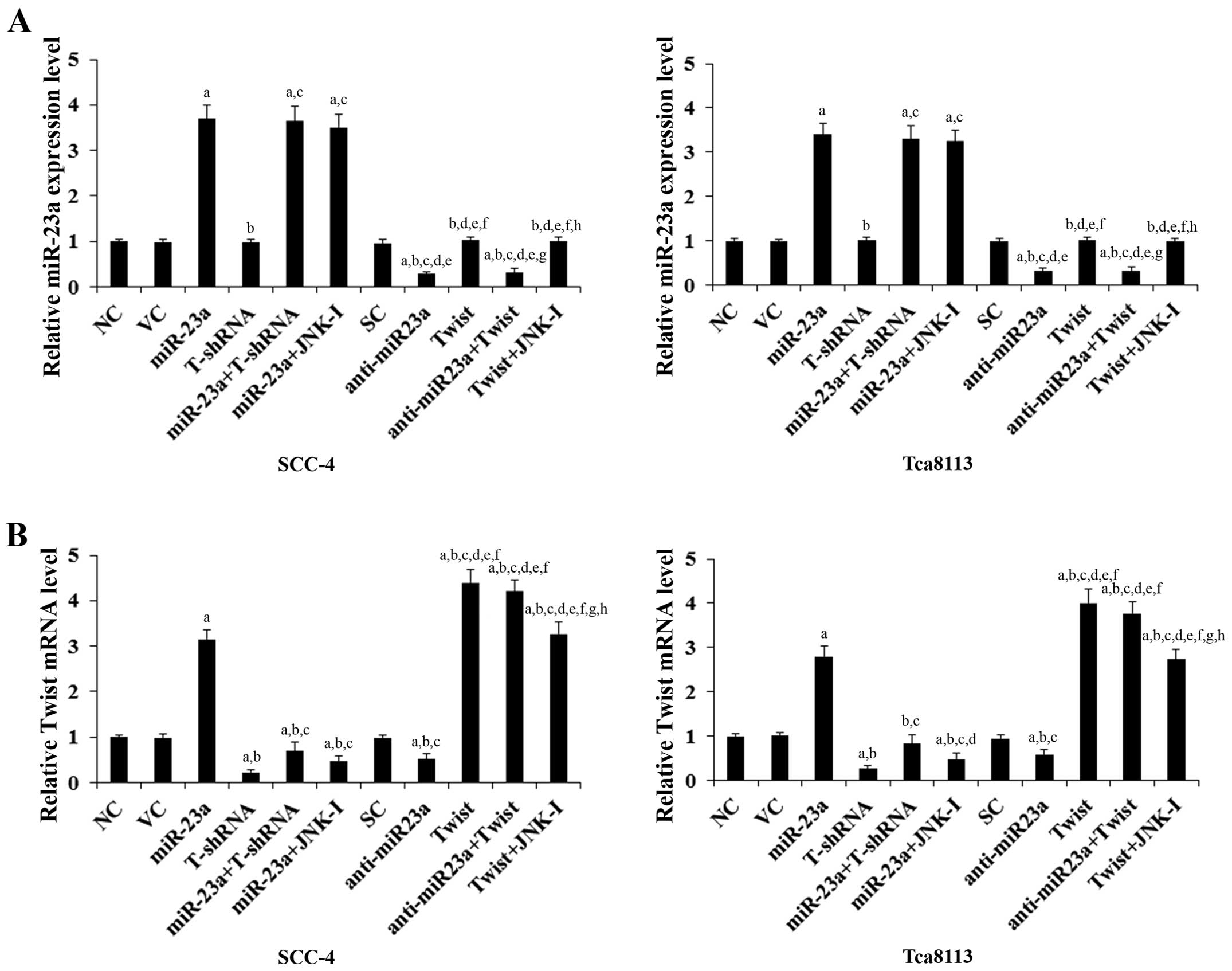 | Figure 1Expression levels of miR-23a and Twist
mRNA in human tongue squamous cell carcinoma (TSCC) cells with
overexpression and knockdown of miR-23a and/or Twist. In SCC-4 and
Tca8113 human TSCC cells, the expression levels of (A) miR-23a and
(B) Twist mRNA were determined by real-time quantitative reverse
transcription PCR in normal control cells (NC), cells transduced
with empty lentiviral vector pMIR and scramble control lentiviral
shRNA (VC), cells overexpressing miR-23a (transduced with
lentiviral pMIRH23aPA-1), cells stably transduced with lentiviral
Twist-shRNA (T-shRNA), cells stably transduced with lentiviral
Twist-shRNA and overexpressing miR-23a (miR-23a+T-shRNA), cells
overexpressing miR-23a and treated with selective c-Jun N-terminal
kinase (JNK) inhibitor SP600125 (10 μM) for 30 min (miR-23a+JNK-I),
cells transduced with lentiviral vector pMIRZIP expressing scramble
control hairpin vector sequences and transfected with empty
pcDNA3.1 vector (SC), cells overexpressing lentiviral shRNA against
miR-23a (anti-miR23a; transduced with lentiviral pMIRZIP-23a),
cells overexpressing Twist (stably transfected with pcDNA3.1-Twist
expression vector), cells overexpressing anti-miR23a and Twist, and
cells overexpressing Twist and treated with SP600125 (10 μM) for 30
min (Twist+JNK-I). The expression level of miR-23a and Twist mRNA
is shown as fold-changes to that of NC (designated as 1). Each
experiment was repeated three times in duplicates. Data values are
expressed as means ± SD. ap<0.05 vs. controls (NC, VC
and SC); bp<0.05 vs. miR-23a; cp<0.05
vs. T-shRNA; dp<0.05 vs. miR-23a+T-shRNA;
ep<0.05 vs. miR-23a+JNK-I; fp<0.05 vs.
anti-miR23a; gp<0.05 vs. Twist; hp<0.05
vs. anti-miR23a+Twist. |
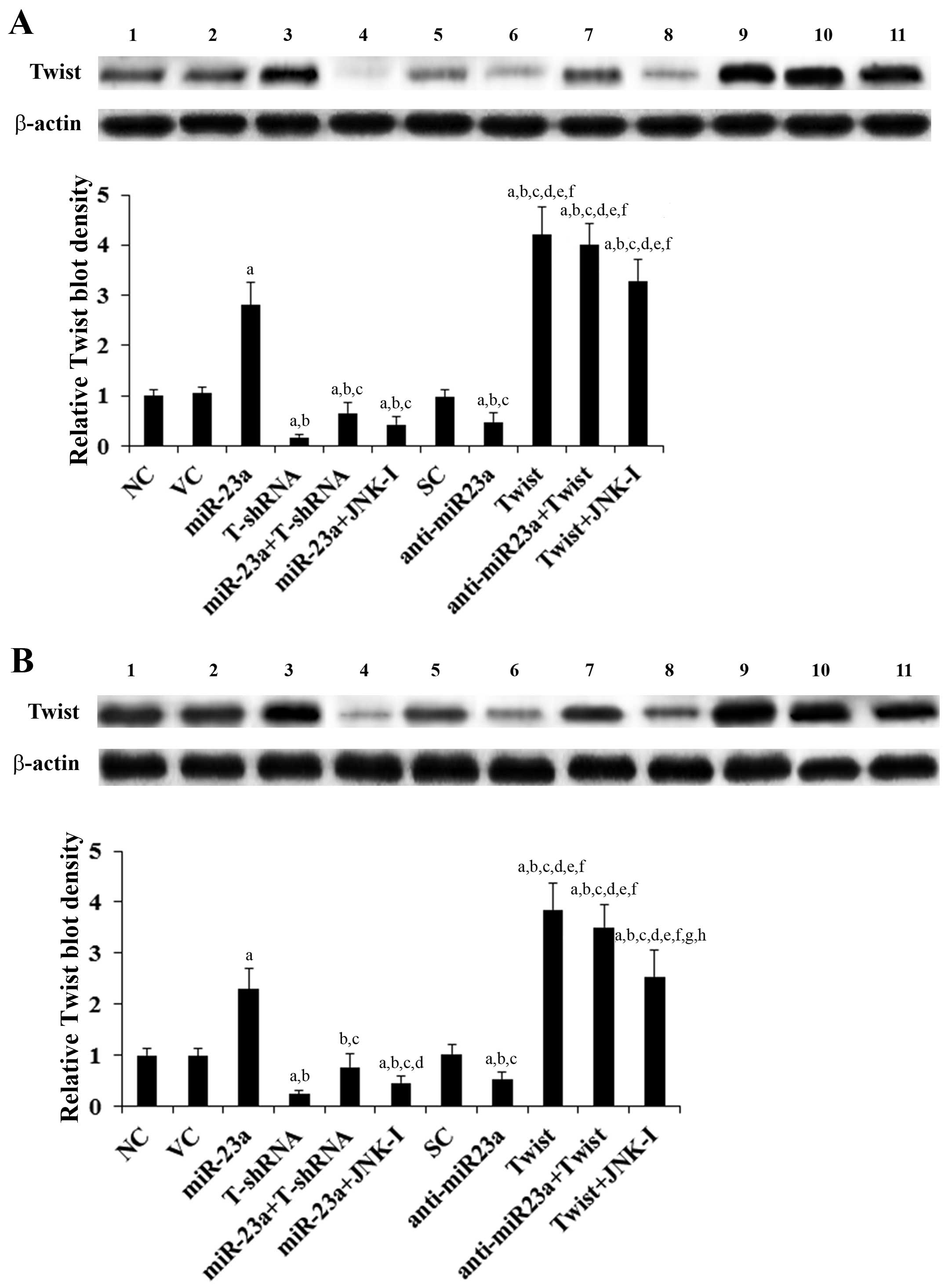 | Figure 2Protein levels of Twist in tongue
squamous cell carcinoma (TSCC) cells with overexpression and
knockdown of miR-23a and/or Twist. In (A) SCC-4 and (B) Tca8113
human TSCC cells, the protein levels of Twist were determined with
western blot analysis in normal control cells (NC, lane 1), cells
transduced with empty lentiviral vector pMIR and scramble control
lentiviral shRNA (VC, lane 2), cells overexpressing miR-23a
(transduced with lentiviral pMIRH23aPA-1) (lane 3), cells stably
transduced with lentiviral Twist-shRNA (T-shRNA, lane 4), cells
stably transduced with lentiviral Twist-shRNA and overexpressing
miR-23a (miR-23a+T-shRNA, lane 5), cells overexpressing miR-23a and
treated with selective c-Jun N-terminal kinase (JNK) inhibitor
SP600125 (10 μM) for 30 min (miR-23a+JNK-I, lane 6), cells
transduced with lentiviral vector pMIRZIP expressing scramble
control hairpin vector sequences and transfected with empty
pcDNA3.1 vector (SC, lane 7), cells overexpressing lentiviral shRNA
against miR-23a (transduced with lentiviral pMIRZIP-23a)
(anti-miR23a, lane 8), cells overexpressing Twist (stably
transfected with pcDNA3.1-Twist expression vector) (lane 9), cells
overexpressing anti-miR23a and Twist (lane 10), and cells
overexpressing Twist and treated with SP600125 (10 μM) for 30 min
(Twist+JNK-I, lane 11). β-actin blotting was used as a loading
control. Density of the Twist blot was normalized against that of
the β-actin blot to obtain a relative blot density, which was
expressed as a fold-change to that of NC (designated as 1). Three
independent experiments were performed for each western blot
analysis. Data values are expressed as means ± SD.
ap<0.05 vs. controls (NC, VC and SC);
bp<0.05 vs. miR-23a; cp<0.05 vs.
T-shRNA; dp<0.05 vs. miR-23a+T-shRNA;
ep<0.05 vs. miR-23a+JNK-I; fp<0.05 vs.
anti-miR23a; gp<0.05 vs. Twist; hp<0.05
vs. anti-miR23a+Twist. |
Effects of overexpression and knockdown
of miR-23a and Twist on JNK activity in TSCC cells
The above results suggested that miR-23a regulates
Twist expression in TSCC cells in a JNK-dependent manner.
Therefore, we next examined the individual effect of and
interaction between miR-23a and Twist on JNK activity, which was
measured by phosphorylation of c-Jun, a substrate of JNK (13). As evidenced by the increased level
of phosphorylated c-Jun, overexpression of miR-23a respectively
induced JNK activity by 2.4- and 2.1-fold in the SCC-4 and Tca8113
cells, which was abolished by SP600125 (10 μM) yet not knockdown of
Twist (Fig. 3). On the other hand,
knockdown of miR-23a decreased JNK activity by ~70% in both SCC-4
and Tca8113 cells, which was not significantly affected by
overexpression of Twist (Fig. 3).
Compared with the controls, overexpression and knockdown of Twist
showed no significant effect on JNK activity (Fig. 3).
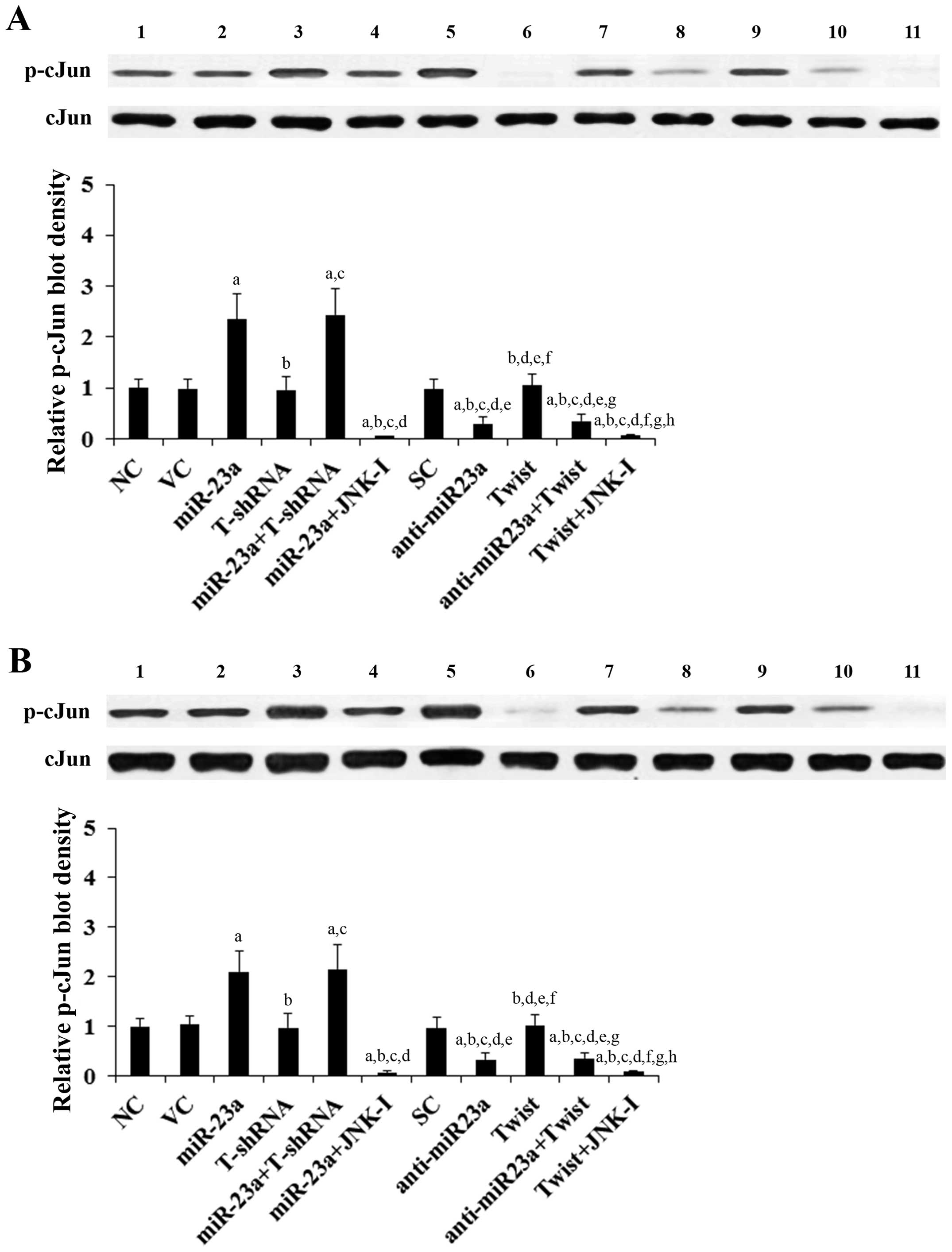 | Figure 3Effect of overexpression and knockdown
of miR-23a and/or Twist on c-Jun N-terminal kinase (JNK) activity
in tongue squamous cell carcinoma (TSCC) cells. In (A) SCC-4 and
(B) Tca8113 TSCC cells, the JNK activity was determined by
measuring phosphorylation of c-Jun, a substrate of JNK. The levels
of phosphorylated c-Jun (p-cJun2) were determined with western blot
analysis in normal control cells (NC, lane 1), cells transduced
with empty lentiviral vector pMIR and scramble control lentiviral
shRNA (VC, lane 2), cells overexpressing miR-23a (transduced with
lentiviral pMIRH23aPA-1) (lane 3), cells stably transduced with
lentiviral Twist-shRNA (T-shRNA, lane 4), cells stably transduced
with lentiviral Twist-shRNA and overexpressing miR-23a
(miR-23a+T-shRNA, lane 5), cells overexpressing miR-23a and treated
with selective c-Jun N-terminal kinase (JNK) inhibitor SP600125 (10
μM) for 30 min (miR-23a+JNK-I, lane 6), cells transduced with
lentiviral vector pMIRZIP expressing scramble control hairpin
vector sequences and transfected with empty pcDNA3.1 vector (SC,
lane 7), cells overexpressing lentiviral shRNA against miR-23a
(transduced with lentiviral pMIRZIP-23a) (anti-miR23a, lane 8),
cells overexpressing Twist (stably transfected with pcDNA3.1-Twist
expression vector) (lane 9), cells overexpressing anti-miR23a and
Twist (lane 10), and cells overexpressing Twist and treated with
SP600125 (10 μM) for 30 min (Twist+JNK-I, lane 11). Density of the
p-cJun blot was normalized against that of c-Jun (cJun) to obtain a
relative blot density, which was expressed as a fold-change to that
of NC (designated as 1) as a measure of the JNK activity. Each
experiment was repeated three times in duplicates. Data values are
expressed as means ± SD. ap<0.05 vs. controls (NC, VC
and SC); bp<0.05 vs. miR-23a; cp<0.05
vs. T-shRNA; dp<0.05 vs. miR-23a+T-shRNA;
ep<0.05 vs. miR-23a+JNK-I; fp<0.05 vs.
anti-miR23a; gp<0.05 vs. Twist; hp<0.05
vs. anti-miR23a+Twist. |
Effects of overexpression and knockdown
of miR-23a and Twist on TSCC cell chemoresistance to cisplatin
To explore the individual effect of and interaction
between miR-23a and Twist on TSCC chemoresistance, we examined
cisplatin IC50 in TSCC cells. A higher IC50
value was considered to correspond to clinical chemoresistance to
cisplatin (16). As shown in
Fig. 4, following 48 h of cisplatin
treatment, the cisplatin IC50 values for SCC-4 and
Tca8113 cells were 6.2 and 7.1 μM, respectively. Overexpression of
miR-23a significantly increased the IC50 to 17.5 and
16.0 μM, respectively, which was abolished by knockdown of Twist or
SP600125 (10 μM) (Fig. 4). In
contrast, knockdown of miR-23a respectively decreased the
IC50 to 3.3 and 4.0 μM, which was completely reversed by
overexpression of Twist (Fig. 4).
Overexpression of Twist respectively increased the cisplatin
IC50 to 22.1 and 20.6 μM in SCC-4 and Tca8113 cells,
while knockdown of Twist decreased the IC50 to 2.6 and
3.3 μM, respectively (Fig. 4).
 | Figure 4Effect of overexpression and knockdown
of miR-23a and/or Twist on cisplatin chemoresistance in tongue
squamous cell carcinoma (TSCC) cells. (A) SCC-4 and (B) Tca8113
TSCC cells were treated with or without various concentrations of
cisplatin for 48 h. The half maximal inhibitory concentration
(IC50) values were determined in normal control cells
(NC), cells transduced with empty lentiviral vector pMIR and
scramble control lentiviral shRNA (VC), cells overexpressing
miR-23a (transduced with lentiviral pMIRH23aPA-1), cells stably
transduced with lentiviral Twist-shRNA (T-shRNA), cells stably
transduced with lentiviral Twist-shRNA and overexpressing miR-23a
(miR-23a+T-shRNA), cells overexpressing miR-23a and treated with
selective c-Jun N-terminal kinase (JNK) inhibitor SP600125 (10 μM)
for 30 min (miR-23a+JNK-I), cells transduced with lentiviral vector
pMIRZIP expressing scramble control hairpin vector sequences and
transfected with empty pcDNA3.1 vector (SC), cells overexpressing
lentiviral shRNA against miR-23a (anti-miR23a; transduced with
lentiviral pMIRZIP-23a), cells overexpressing Twist (stably
transfected with pcDNA3.1-Twist expression vector), cells
overexpressing anti-miR23a and Twist, and cells overexpressing
Twist and treated with SP600125 (10 μM) for 30 min (Twist+JNK-I).
Each experiment was repeated three times in duplicates. Data values
are expressed as means ± SD. ap<0.05 vs. controls
(NC, VC and SC); bp<0.05 vs. miR-23a;
cp<0.05 vs. T-shRNA; dp<0.05 vs.
miR-23a+T-shRNA; ep<0.05 vs. miR-23a+JNK-I;
fp<0.05 vs. anti-miR23a. |
Effects of overexpression and knockdown
of miR-23a and Twist on cisplatin-induced apoptosis in TSCC
cells
We next examined the individual effect of and
interaction between miR-23a and Twist on cisplatin-induced
apoptosis in the TSCC cells. Under normal culture conditions,
overexpression and knockdown of miR-23a and Twist showed no
significant effect on TSCC cell apoptosis compared with the
controls (data not shown). After 30 h of cisplatin (20 μM)
treatment, the percentages of apoptotic cells in SCC-4 (Fig. 5) and Tca8113 (Fig. 6) cells were 20.1 and 22.7%,
respectively. Overexpression of miR-23a significantly decreased
cell apoptosis to 8.3 and 11.9%, respectively, which was completely
reversed by knockdown of Twist or by SP600125 (10 μM) (Figs. 5 and 6). In contrast, knockdown of miR-23a
respectively increased cell apoptosis to 31.2 and 34.0%, which was
abolished by overexpression of Twist (Figs. 5 and 6). Overexpression of Twist decreased
apoptosis of SCC-4 (Fig. 5) and
Tca8113 (Fig. 6) cells to 6.7 and
8.1%, respectively, while knockdown of Twist increased cell
apoptosis to 41.4 and 39.6%, respectively.
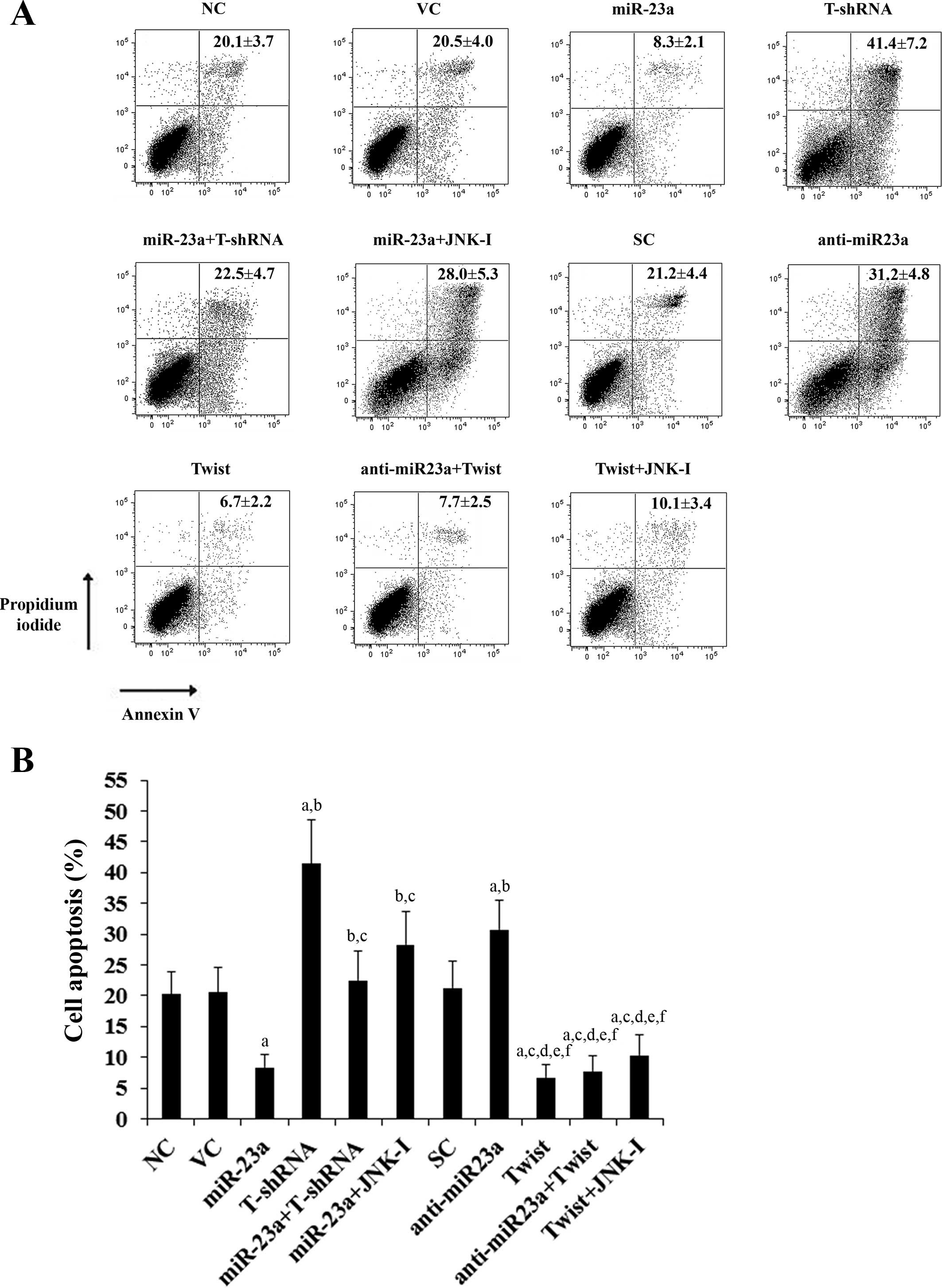 | Figure 5Effect of overexpression and knockdown
of miR-23a and/or Twist on apoptosis of SCC-4 tongue squamous cell
carcinoma (TSCC) cells. SCC-4 TSCC cells were treated with 20 μM of
cisplatin for 30 h. (A) Cell apoptosis was quantitated using
Annexin V/propidium iodide staining coupled with flow cytometric
analysis in normal control cells (NC), cells transduced with empty
lentiviral vector pMIR and scramble control lentiviral shRNA (VC),
cells overexpressing miR-23a (transduced with lentiviral
pMIRH23aPA-1), cells stably transduced with lentiviral Twist-shRNA
(T-shRNA), cells stably transduced with lentiviral Twist-shRNA and
overexpressing miR-23a (miR-23a+T-shRNA), cells overexpressing
miR-23a and treated with selective c-Jun N-terminal kinase (JNK)
inhibitor SP600125 (10 μM) for 30 min (miR-23a+JNK-I), cells
transduced with lentiviral vector pMIRZIP expressing scramble
control hairpin vector sequences and transfected with empty
pcDNA3.1 vector (SC), cells overexpressing lentiviral shRNA against
miR-23a (anti-miR23a; transduced with lentiviral pMIRZIP-23a),
cells overexpressing Twist (stably transfected with pcDNA3.1-Twist
expression vector), cells overexpressing anti-miR23a and Twist, and
cells overexpressing Twist and treated with SP600125 (10 μM) for 30
min (Twist+JNK-I). Each experiment was repeated three times in
duplicates. Sums of percentages of early apoptotic cells (left
lower quadrant) and late apoptotic cells/dead cells (right upper
quadrant) are expressed as means ± SD in the right upper quadrant
of each dot figure. (B) Sums of the percentages of early apoptotic
cells (left lower quadrant) and late apoptotic cells/dead cells
(right upper quadrant) are shown as percentages of cell apoptosis
in the histograms (means ± SD). ap<0.05 vs. controls
(NC, VC and SC); bp<0.05 vs. miR-23a;
cp<0.05 vs. T-shRNA; dp<0.05 vs.
miR-23a+T-shRNA; ep<0.05 vs. miR-23a+JNK-I;
fp<0.05 vs. anti-miR23a. |
 | Figure 6Effect of overexpression and knockdown
of miR-23a and/or Twist on apoptosis of Tca8113 tongue squamous
cell carcinoma (TSCC) cells. Tca8113 TSCC cells were treated with
20 μM of cisplatin for 30 h. (A) Cell apoptosis was quantitated
using Annexin V/propidium iodide staining coupled with flow
cytometric analysis in normal control cells (NC), cells transduced
with empty lentiviral vector pMIR and scramble control lentiviral
shRNA (VC), cells overexpressing miR-23a (transduced with
lentiviral pMIRH23aPA-1), cells stably transduced with lentiviral
Twist-shRNA (T-shRNA), cells stably transduced with lentiviral
Twist-shRNA and overexpressing miR-23a (miR-23a+T-shRNA), cells
overexpressing miR-23a and treated with selective c-Jun N-terminal
kinase (JNK) inhibitor SP600125 (10 μM) for 30 min (miR-23a+JNK-I),
cells transduced with lentiviral vector pMIRZIP expressing scramble
control hairpin vector sequences and transfected with empty
pcDNA3.1 vector (SC), cells overexpressing lentiviral shRNA against
miR-23a (anti-miR23a; transduced with lentiviral pMIRZIP-23a),
cells overexpressing Twist (stably transfected with pcDNA3.1-Twist
expression vector), cells overexpressing anti-miR23a and Twist, and
cells overexpressing Twist and treated with SP600125 (10 μM) for 30
min (Twist+JNK-I). Each experiment was repeated three times in
duplicates. Sums of the percentages of early apoptotic cells (left
lower quadrant) and late apoptotic cells/dead cells (right upper
quadrant) are expressed as means ± SD in the right upper quadrant
of each dot figure. (B) Sums of the percentages of early apoptotic
cells (left lower quadrant) and late apoptotic cells/dead cells
(right upper quadrant) are shown as percentages of cell apoptosis
in the histogram (means ± SD). ap<0.05 vs. controls
(NC, VC and SC); bp<0.05 vs. miR-23a;
cp<0.05 vs. T-shRNA; dp<0.05 vs.
miR-23a+T-shRNA; ep<0.05 vs. miR-23a+JNK-I;
fp<0.05 vs. anti-miR23a. |
Discussion
TSCC is associated with poorer survival and a lower
rate of local tumor control than other sites of head and neck
cancer (1). Innate or acquired
resistance to cisplatin, a standard chemotherapy agent for TSCC, is
common in patients with TSCC (4–6).
Increased expression of miR-23a reportedly promotes cisplatin
chemoresistance in TSCC cells (3).
High expression of Twist is also associated with cancer
chemoresistance (10) and poor
prognosis of TSCC patients (11).
In the present study, we demonstrate that miR-23a promotes
cisplatin chemoresistance in TSCC cells largely through Twist.
We employed cisplatin IC50 as a measure
of cisplatin chemoresistance in TSCC cells. A higher
IC50 value was considered to correspond to clinical
chemoresistance to cisplatin (16).
As evidenced by overexpression and knockdown experiments, miR-23a
and Twist individually increased cisplatin
IC50/cisplatin chemoresistance in TSCC cells. Knockdown
of Twist abolished the chemoresistance-promoting effect of
overexpressing miR-23a, while overexpression of Twist completely
reversed the inhibitory effect of knocking down miR-23a on
cisplatin chemoresistance. The findings indicate that Twist is
functionally downstream of miR-23a and largely mediates the
promoting effects of miR-23a on cisplatin chemoresistance in TSCC
cells, which corroborates our finding that miR-23a induces Twist
expression in TSCC cells. In addition, miR-23a and Twist
individually showed a protective effect against cisplatin-induced
apoptosis in TSCC cells. Knockdown of Twist abolished the
protective effect of overexpressing miR-23a, while overexpression
of Twist completely reversed the apoptosis-enhancing effect of
knocking down miR-23a. The protective effect of miR-23a/Twist
signaling against cisplatin-induced apoptosis serves as a
mechanistic explanation for miR-23a/Twist-induced cisplatin
chemoresistance in TSCC cells.
Overexpression and knockdown of miR-23a in TSCC
cells respectively increased and decreased the expression of Twist
at both the mRNA and the protein levels, yet not vice versa. In
addition, a selective JNK inhibitor readily abolished
miR-23a-induced Twist expression in TSCC cells without
significantly altering the expression of miR-23a, indicating that
miR-23a induces Twist expression in a JNK-dependent manner in TSCC
cells. How miR-23a induces the expression of Twist through JNK in
TSCC cells will be explored by us in subsequent studies.
While selective JNK inhibitor SP600125 cancelled the
promoting effects of miR-23a on cisplatin chemoresistance in TSCC
cells, overexpression of Twist still significantly augmented
cisplatin chemoresistance in the presence of SP600125. The results
suggest that miR-23a and Twist act functionally upstream and
downstream of JNK, respectively. This is in agreement with our
findings that while overexpression and knockdown of miR-23a
respectively increased and decreased JNK activity, Twist showed no
significant effect on JNK activity in TSCC cells. This is also in
agreement with a previous report that JNK signaling may control the
expression of twist (17). Previous
studies have suggested that the JNK signaling pathway is clearly a
basis for resistance to DNA-damaging drugs (18). Our findings indicate that JNK
mediates miR-23a-induced Twist expression in TSCC cells, which
markedly promotes cisplatin chemoresistance. Therefore, the
functional role of JNK signaling in cisplatin chemoresistance in
TSCC cells is at least partially fulfilled through miR-23a/Twist
signaling.
Yu et al demonstrated that miR-23a functions
as an upstream regulator of DNA topoisomerase IIβ to induce
cisplatin chemo-resistance in TSCC cells (3). In the present study, we showed that
miR-23a promotes cisplatin chemoresistance in TSCC cells through
Twist. Thus, whether and how TOP2B and Twist would interact to
impact cisplatin chemoresistance in TSCC cells would be a
significant topic for future studies. Cisplatin elicits DNA repair
mechanisms by crosslinking DNA, which in turn activates apoptosis
when repair proves impossible (19). In the present study, we only
examined the effect of miR-23a/Twist signaling on cisplatin
chemoresistance in TSCC cells. It is unclear whether miR-23a/Twist
would impact chemoresistance to other types of chemotherapy agents.
Further studies with more types of chemotherapy agents and TSCC
cell lines would elucidate this issue. In addition, since Twist has
been associated with chemoresistance in a variety of cancers such
as nasopharyngeal, breast, prostate and lung cancer (10,20),
it is worth defining the role of miR-23a/Twist signaling in other
cancer types besides TSCC in future studies.
In conclusion, the present study for the first time
demonstrated that miR-23a promotes cisplatin chemoresistance and
protects against cisplatin-induced apoptosis in TSCC cells through
inducing Twist expression by a JNK-dependent mechanism. It adds new
insights into the molecular mechanisms underlying TSCC
chemoresistance.
Acknowledgements
This study was supported by the Science and
Technology Bureau of Hunan Province (grant no. 2013-SK-3203).
References
|
1
|
Choi KK, Kim MJ, Yun PY, Lee JH, Moon HS,
Lee TR and Myoung H: Independent prognostic factors of 861 cases of
oral squamous cell carcinoma in Korean adults. Oral Oncol.
42:208–217. 2006. View Article : Google Scholar
|
|
2
|
Xing Y, Qi J, Deng S, Wang C, Zhang L and
Chen J: Small interfering RNA targeting ILK inhibits metastasis in
human tongue cancer cells through repression of
epithelial-to-mesenchymal transition. Exp Cell Res. 319:2058–2072.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu ZW, Zhong LP, Ji T, Zhang P, Chen WT
and Zhang CP: MicroRNAs contribute to the chemoresistance of
cisplatin in tongue squamous cell carcinoma lines. Oral Oncol.
46:317–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Price KA and Cohen EE: Current treatment
options for metastatic head and neck cancer. Curr Treat Options
Oncol. 13:35–46. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wingo PA, Tong T and Bolden S: Cancer
statistics, 1995. CA Cancer J Clin. 45:8–30. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gu Y, Fan S, Xiong Y, Peng B, Zheng G, Yu
Y, Ouyang Y and He Z: Cloning and functional characterization of
TCRP1, a novel gene mediating resistance to cisplatin in an oral
squamous cell carcinoma cell line. FEBS Lett. 585:881–887. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feng B, Wang R and Chen LB: Review of
miR-200b and cancer chemosensitivity. Biomed Pharmacother.
66:397–402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Filipowicz W, Jaskiewicz L, Kolb FA and
Pillai RS: Post-transcriptional gene silencing by siRNAs and
miRNAs. Curr Opin Struct Biol. 15:331–341. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ansieau S, Morel AP, Hinkal G, Bastid J
and Puisieux A: TWISTing an embryonic transcription factor into an
oncoprotein. Oncogene. 29:3173–3184. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
da Silva SD, Alaoui-Jamali MA, Soares FA,
Carraro DM, Brentani HP, Hier M, Rogatto SR and Kowalski LP: TWIST1
is a molecular marker for a poor prognosis in oral cancer and
represents a potential therapeutic target. Cancer. 120:352–362.
2014. View Article : Google Scholar
|
|
12
|
Matsuo N, Shiraha H, Fujikawa T, Takaoka
N, Ueda N, Tanaka S, Nishina S, Nakanishi Y, Uemura M, Takaki A,
Nakamura S, Kobayashi Y, Nouso K, Yagi T and Yamamoto K: Twist
expression promotes migration and invasion in hepatocellular
carcinoma. BMC Cancer. 9:2402009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Muniyappa H and Das KC: Activation of
c-Jun N-terminal kinase (JNK) by widely used specific p38 MAPK
inhibitors SB202190 and SB203580: a MLK-3-MKK7-dependent mechanism.
Cell Signal. 20:675–683. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ding X, Zhang Z, Li S and Wang A:
Combretastatin A4 phosphate induces programmed cell death in
vascular endothelial cells. Oncol Res. 19:303–309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Venugopal SK, Chen J, Zhang Y, Clemens D,
Follenzi A and Zern MA: Role of MAPK phosphatase-1 in sustained
activation of JNK during ethanol-induced apoptosis in
hepatocyte-like VL-17A cells. J Biol Chem. 282:31900–31908. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu M, Wang J, Huang H, Hou J, Zhang B and
Wang A: miR-181a-Twist1 pathway in the chemoresistance of tongue
squamous cell carcinoma. Biochem Biophys Res Commun. 441:364–370.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhan X, Feng X, Kong Y, Chen Y and Tan W:
JNK signaling maintains the mesenchymal properties of multi-drug
resistant human epidermoid carcinoma KB cells through snail and
twist1. BMC Cancer. 13:1802013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vasilevskaya I and O’Dwyer PJ: Role of Jun
and Jun kinase in resistance of cancer cells to therapy. Drug
Resist Updat. 6:147–156. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rosenberg B, VanCamp L, Trosko JE and
Mansour VH: Platinum compounds: a new class of potent antitumour
agents. Nature. 222:385–386. 1969. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Entz-Werlé N, Choquet P, Neuville A,
Kuchler-Bopp S, Clauss F, Danse JM, Simo-Noumbissie P, Guérin E,
Gaub MP, Freund JN, Boehm N, Constantinesco A, Lutz P, Guenot D and
Perrin-Schmitt F: Targeted Apc;Twist double-mutant mice: a new
model of spontaneous osteosarcoma that mimics the human disease.
Transl Oncol. 3:344–353. 2010. View Article : Google Scholar : PubMed/NCBI
|














