Introduction
Over half the cases of hepatocellular carcinomas
(HCCs) are found to be inoperable by curative treatments such as
surgery and radiofrequency ablation (RFA). Alternatively,
transarterial chemoembolization (TACE) and sorafenib therapy are
ideal palliative treatment options for HCC (1). However, TACE or sorafenib therapy
alone rarely achieves a complete or satisfactory response.
Therefore, a combination of additional treatments, such as
radiotherapy (RT), with conventional ones are under consideration.
Recent studies reported RT as a salvage treatment option for HCCs
that are difficult to operate by TACE (2–6).
Irradiation induces pro-inflammatory signaling
associated with anti-apoptosis, proliferation, angiogenesis and
invasiveness, which are mediated through the activation of nuclear
factor-κB (NF-κB) (7). The
pro-survival pathways impart radioresistance to tumor cells. In
addition, hypoxia inhibits the repair of DNA damage caused by
irradiation and induces several signaling factors such as hypoxia
inducible factor-1α (HIF-1α), resulting in the development of
radioresistance (8,9). HCC is frequently exposed to hypoxia
due to rapid cell growth. Moreover, TACE or sorafenib can be used
to produce a hypoxic environment via embolization of the feeding
artery or anti-angiogenesis.
Emodin (1,3,8-trihydroxy-6-methylanthraquinone), a
plant-derived polyphenol, has been reported to possess anti-cancer
properties (10). It was previously
reported that emodin inhibits cell growth by suppressing NF-κB and
increases apoptosis in human HCC cell lines (11–13).
Other studies reported that emodin inhibits hypoxia-induced
signaling factors, such as HIF-1α, in several cell lines (10,14,15).
However, the data regarding the role of emodin as a
radiosensitizer in human HCC cell line are limited. Therefore, in
this study, we investigated whether emodin attenuates
hypoxia-induced radioresistance in the HepG2 human HCC cell line as
well as the underlying mechanism of radiosensitization.
Materials and methods
Cell culture and treatment
The HepG2 human HCC cell line was obtained from the
American Type Culture Collection (Rockville, MD, USA) and
maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Welgene,
Daegu, Korea) supplemented with 1 mM sodium pyruvate, 10% fetal
bovine serum (FBS; HyClone, Logan, UT, USA), and 2%
penicillin/streptomycin (Gibco, Carlsbad, CA, USA). The cells were
cultured at 37°C under a humidified atmosphere of 5%
CO2. The media were supplemented with fresh media every
3 days. The cells were maintained under hypoxia in a glove box-type
anaerobic chamber (Thermo Forma, Marietta, OH, USA). Hypoxia was
created by maintaining the gas composition at <1% O2,
5% CO2, 10% H2, and 85% N2 (under
continuous computerized monitoring), indicating a partial oxygen
pressure of <15 mmHg at 37°C. Oxygen-dependent experiments were
performed in hypoxic and normoxic incubators.
Irradiation and emodin treatment
Overnight cells incubated at 37°C were exposed to
normoxia (20% O2) or hypoxia (1% O2) for 24
h. The cells were then exposed to 10 μM emodin under normoxia for
24 h, followed by exposure to gamma-rays from a
137Cs-ray source (Eckert & Ziegler, Berlin, Germany)
at a dose rate of 2.6 Gy/min. Following irradiation with 10-Gy
dose, the cells were incubated under normoxia or hypoxia at 37°C
(Fig. 1B).
Antibodies and reagents
The antibody against poly(ADP-ribose) polymerase 1
(PARP1) was obtained from Santa Cruz Biotechnology, Inc. (Mouse;
1:1,000; Santa Cruz, CA, USA). JMJD1A and JMJD2B antibodies were
purchased from Abcam (Cambridge, UK). HIF-1α antibody was purchased
from Novus Biologicals (Littelton, CO, USA). Anti-β-actin antibody
was purchased from Sigma-Aldrich (St. Louis, MO, USA) and was
incubated with specific horseradish peroxidase-conjugated secondary
antibodies (Invitrogen, Carlsbad, CA, USA). Emodin was purchased
from LC Laboratories (Woburn, MA, USA) and solubilized in DMSO.
DMSO was used in all the experiments as a vehicle control.
Cell proliferation assay
The 3-(4,5-dimethylthiazol-2-yl)-
2,5-diphenyltetrazolium bromide (MTT) assay, which is based on the
conversion of MTT to MTT-formazan by mitochondria, was conducted.
HepG2 cells were resuspended and plated in 96-well plates at
1×104 cells/200 μl concentration in culture media
supplemented with 5% FBS and incubated with or without drugs for
24–72 h, followed by incubation with MTT (5 mg/ml in
phosphate-buffered saline; PBS) for 3 h. The plate was then
centrifuged at 2,000 rpm for 5 min at 4°C, and the MTT solution was
removed from the wells by aspiration. Formazan crystals were
dissolved in 2 ml of DMSO. The absorbance was recorded on the
Paradigm Detection Platform (Beckman Coulter, Inc., Fullerton, CA,
USA) at a wavelength of 540 nm.
Cell cycle analysis
The cells were exposed to 10 μg/ml emodin or 10 Gy
radiation for 24 h and then harvested. The harvested cells were
trypsinized, resuspended in 3 ml PBS, centrifuged, and washed with
3 ml PBS. The cells were then fixed in 70% ethanol for 16 h at
−20°C and stained with propidium iodide (PI, 40 μg/ml) and RNAse A
(50 μg/ml). The stained cells were subjected to cell cycle analysis
by using the FACSAria (BD Biosciences, San Jose, CA, USA).
Apoptosis analysis
The Annexin V/PE Apoptosis Detection kit (BD
Biosciences, Bedford, MA, USA) was used to assess Annexin
V-positive cells. Briefly, fresh cell preparations were incubated
with 1X Annexin binding buffer, Annexin V/PE (2.5 μg/ml)-conjugated
primary antibody, and 7-amino actinomycin D (7-AAD) (5 μl) for 15
min in an ice bath. After incubation, 10 μg/ml of PI was added to
the cells, and the cells were analyzed by FACSAria.
Western blotting
The cells were collected with ice-cold PBS and
re-suspended in lysis buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl,
1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium
pyrophosphate, 1 mM β-glycerophosphate, 1 mM
Na3VO4, 1 μg/ml leupeptin, and 1 mM
phenylmethanesulfonyl fluoride]. The suspension was diluted with a
mixture of lithium dodecyl sulfate (LDS) sample buffer and heated
at 95°C for 5 min. The samples were electrophoresed on 10% sodium
dodecyl sulfate-polyacrylamide gels (Invitrogen) and transferred
onto nitrocellulose membranes (GE Healthcare Life Sciences,
Piscataway, NJ, USA). The blots were saturated in TBS-T buffer (20
mM Tris, 137 mM NaCl, 0.05% Tween-20; pH 7.6) containing 3% bovine
serum albumin (BSA) for 1 h at room temperature and then incubated
overnight at 4°C with primary antibodies. The immunoreactive
proteins were detected by enhanced chemiluminescence (Thermo
Scientific, Rockford, IL, USA). The immunoblots were quantified by
the ImageMaster densitometry program.
Statistical analysis
Paired Student’s t-test and Microsoft Excel were
used to assess the data obtained from MTT assays, cell
proliferation, mammosphere formation, and FACSAria, which were
conducted in triplicate and repeated three times. The percentage
inhibition of the western blot data was determined from the ratio
of band density. P<0.05 was considered statistically
significant.
Results
Emodin and radiation additively inhibit
HCC cell growth
To investigate the effect of emodin on cell growth
or viability of HepG2, we treated HepG2 cells with emodin for 24
and 72 h and measured the cell viability by the MTT assay. The
viability of cells treated with emodin was decreased in a
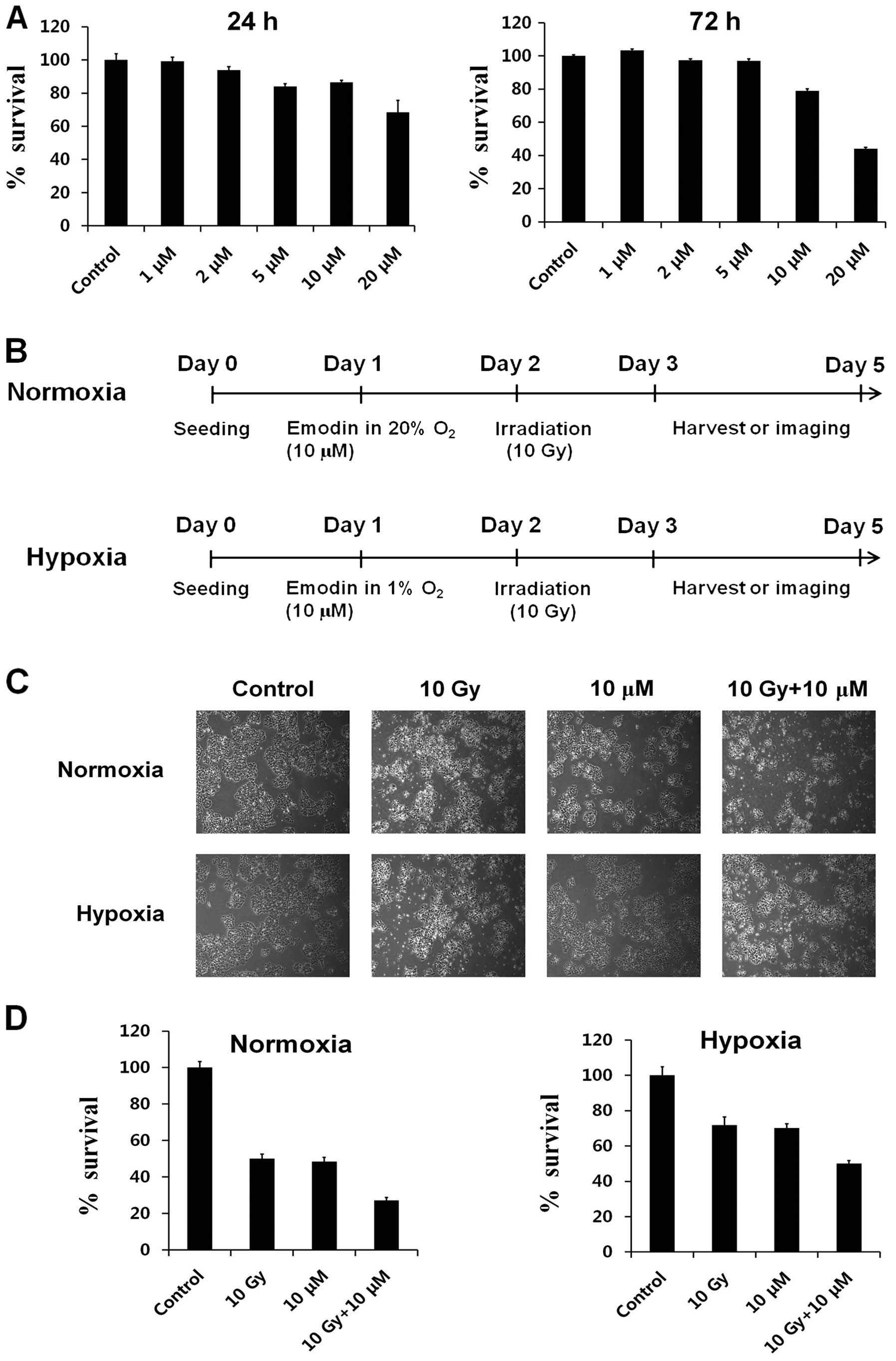
dose-dependent manner (Fig. 1A).
The viability of HepG2 cells decreased to 80% following treatment
with 10 μM emodin. Hypoxia is known to hinder effective RT in
cancer (8,9). Therefore, we investigated whether
emodin increases the radiosensitivity of HepG2 cells after
irradiation under both normoxia and hypoxia (Fig. 1B). For this, we first investigated
the effects of radiation and/or emodin treatment on the
morphological changes under the two environmental conditions. The
density of HepG2 cells under normoxia was decreased by irradiation
or emodin after 72 h of treatment (lanes 2 and 3) under microscopy
(Fig. 1C, upper panel). Moreover,
the combination treatment with radiation and emodin induced
significant decreases in HepG2 cell density (Fig. 1C, lane 4). However, the density of
HepG2 cells decreased less after radiation or emodin treatment
under hypoxia as compared to under normoxia (Fig. 1C, lower panel).
We also analyzed the effects of radiation and emodin
treatment on cell viability using the MTT assay. The cell viability
of HepG2 cells exposed to radiation and/or emodin for 24 h did not
change compared to that in the control group (data not shown).
However, changes were observed in the cell viability of HepG2 cells
after radiation and emodin treatment for 72 h (Fig. 1D, left panel, lane 1 vs. lanes 2 and
3) under normoxia. In addition, we observed a synergistic effect of
the combination of radiation and emodin on HepG2 cell death
(Fig. 1D, left panel, lane 1 vs.
lane 4). Under hypoxia, the viability of HepG2 cells was minimally
decreased after exposure to radiation and/or emodin for 72 h
(Fig. 1D, right panel, lane 1 vs.
lanes 2 and 3). On the other hand, the combination treatment with
radiation and emodin induced a significant decrease in HepG2 cell
viability (Fig. 1D, right panel,
lane 1 vs. lane 4). These results suggested that cancer cell
survival during RT under hypoxia may decrease significantly by
co-treatment of cells with emodin.
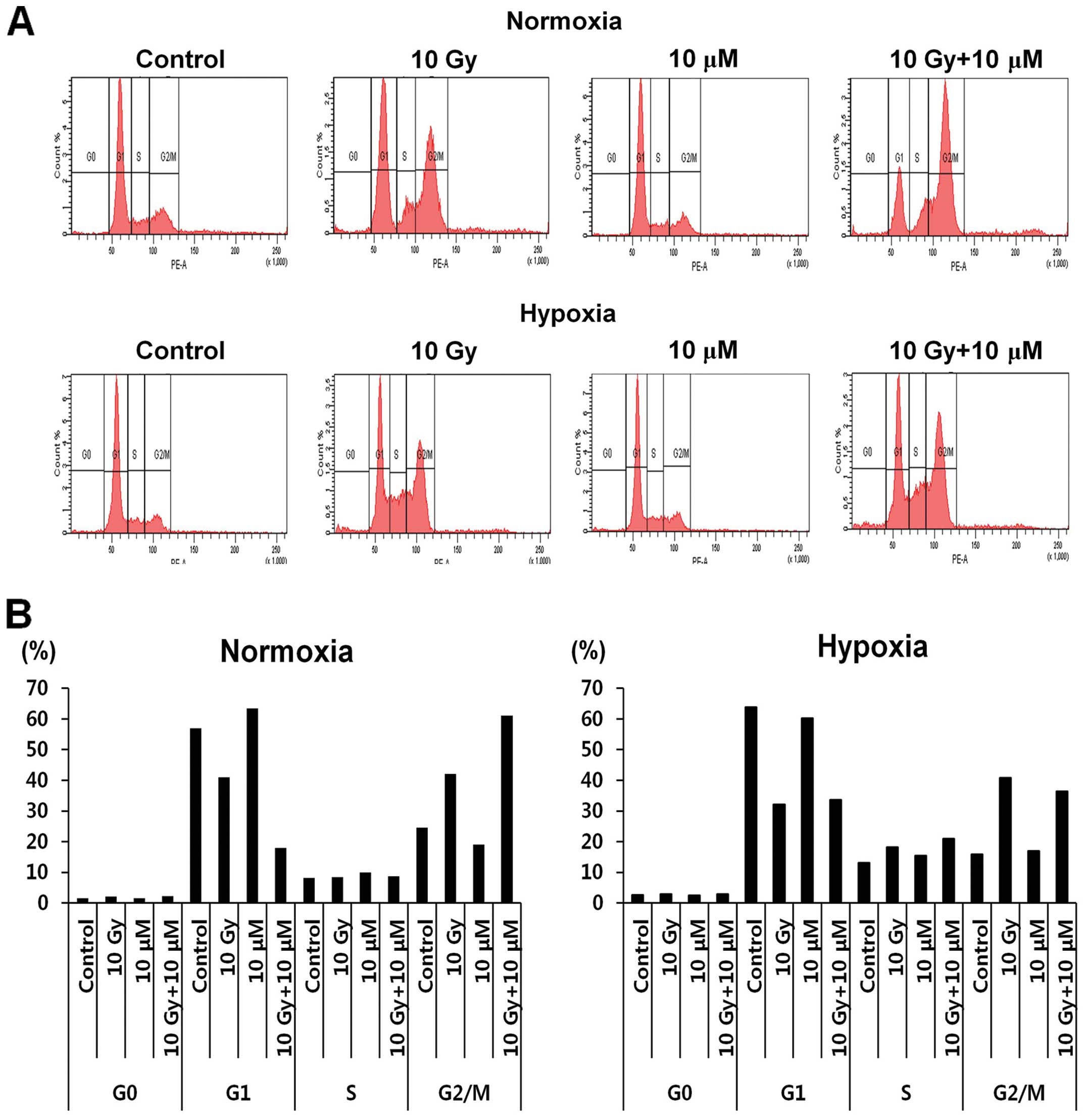
Emodin and radiation modulate HCC cell
cycle progression
Radiation and emodin treatments are known to
modulate several biological processes such as cell death,
proliferation, and differentiation in various cancer types
(10,14,15).
Therefore, we investigated the cell cycle changes during these
treatments to identify the possible action mechanism of these
agents. The results for irradiation treatment (left panel, 10 Gy)
showed arrest of more populations of HepG2 cells in the G2/M phase
than was observed for the control conditions under normoxia
(Fig. 2A and B). The cells treated
with emodin (10 μM) showed a pattern similar to that of the
untreated control cells (Fig. 2,
left panel). Of note, the G2/M population of HepG2 cells in the
combination treatment group [radiation (10 Gy) + emodin (10 μM)]
showed a greater increase than those in the radiation treatment (10
Gy) group (Fig. 2, left panel).
Under hypoxia, emodin treatment showed a similar effect on the cell
cycle regulation of HepG2 cells as that by control treatment,
except that the G2/M population was slightly decreased (Fig. 2, left panel G2/M vs. right panel
G2/M).
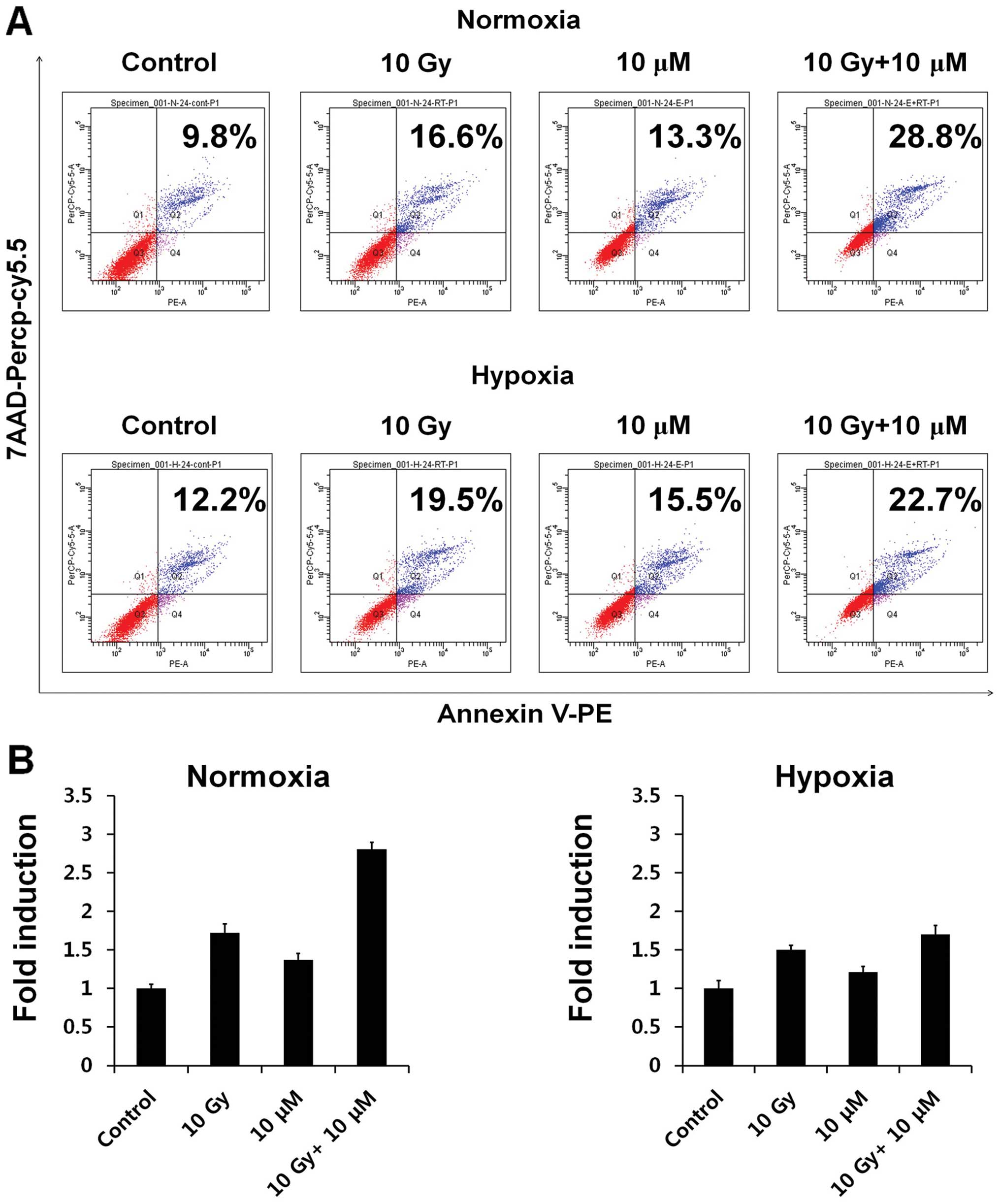
Emodin and radiation induce HCC cell
apoptosis
As viability of HepG2 cells was decreased by
radiation and emodin (Fig. 1D), we
assessed the apoptotic populations by Annexin V/PE staining after
radiation and emodin treatment (Fig. 3A
and B). Under normoxia, the apoptotic population of HepG2 cells
was slightly increased 24 h after emodin treatment (Fig. 3A, upper panel; 10 μM, 9.8 vs.
13.3%). By contrast, compared to the findings for the control,
irradiation was found to significantly stimulate the apoptotic
populations (Fig. 3A, upper panel;
10 Gy, 9.8 vs. 16.6%).
Unlike the results for the control, the combination
treatment with radiation and emodin (10 Gy + 10 μM) showed a
synergistic effect in increasing the apoptotic population of HepG2
cells (Fig. 3A, upper panel; 9.8
vs. 28.8%). This result is consistent with previous cell viability
results (Fig. 1D). In addition, the
combination treatment increased the apoptotic population of HepG2
cells to a greater extent than the control treatment under hypoxia
(Fig. 3B, lower panel; 10 Gy + 10
μM, 12.2 vs. 22.7%), albeit to a lesser extent than that under
normoxia.
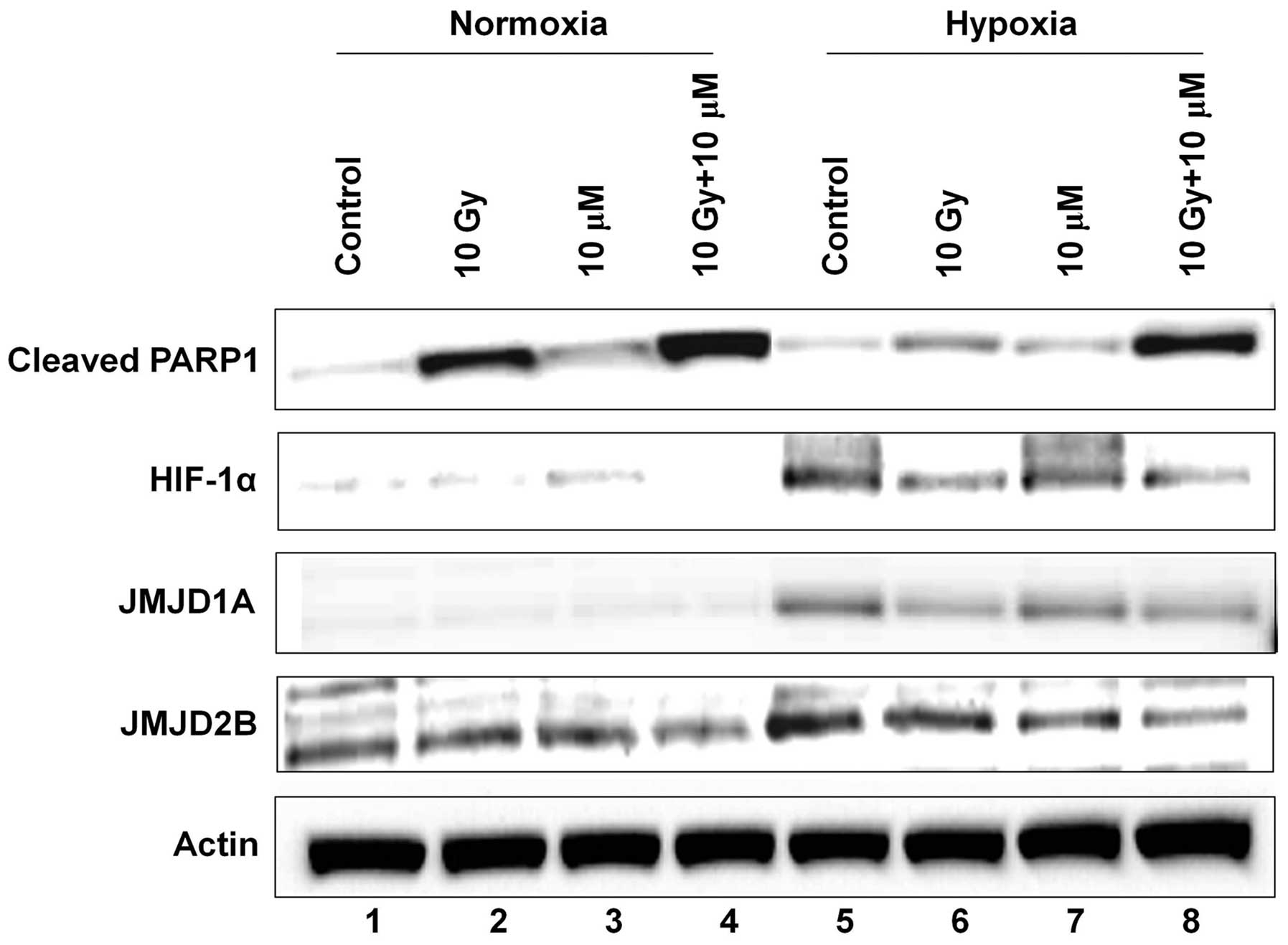
Emodin and radiation induce the
upregulation of cleaved PARP1 and downregulation of JMJD1A and
JMJD2B
To investigate the potential factors that can be
regulated to modify the cell cycle and death of hepatoma cells by
radiation and emodin exposure, we determined the level of cleaved
PARP1 protein by western blotting. Cleaved PARP1 is a well-known
indicator of cell apoptosis. The level of cleaved PARP1 usually
increases by radiation and/or genotoxic reagents. The level of
cleaved PARP1 was significantly increased by irradiation (lane 1
vs. lane 2), whereas it was only minimally increased by emodin
treatment under normoxia (Fig. 4,
lane 1 vs. lane 3). Moreover, combination treatment of HepG2 cells
with radiation and emodin maximized the level of cleaved PARP1
(lane 1 vs. lane 4). Under hypoxia, the level of cleaved PARP1 was
minimally increased by irradiation or emodin treatment. However, a
significant increase was observed in the level of cleaved PARP1
after the combination treatment (lane 5 vs. lane 8).
Hypoxia induces HIF-1α-mediated biological processes
in cancer cells (9,14,16,17).
Therefore, we measured the level of HIF-1α and the expression of
its target genes, JMJD1A and JMJD2B. JMJD1A and
JMJD2B are known to regulate the cell cycle and cell proliferation
under hypoxia (16,17). The levels of HIF-1α, JMJD1A, and
JMJD2B were minimally detected under normoxia (lanes 1–4), whereas
these levels were clearly detected under hypoxia (lanes 5–8).
Radiation downregulated the levels of HIF-1α, JMJD1A, and JMJD2B
(lane 5 vs. lane 6), whereas emodin downregulated only JMJD2B
levels (lane 5 vs. lane 7). Of note, the expression of HIF-1α,
JMJD1A, and JMJD2B significantly decreased after the cells were
exposed to the combination treatment (lane 5 vs. lane 8).
These results suggested that emodin may be crucial
in HCC regulation and that HIF-1α, JMJD1A, and JMJD2B may
constitute a novel therapeutic target to overcome hypoxia-induced
radioresistance, thereby improving the efficiency of RT.
Discussion
HCC is the fifth-most common malignancy and causes
one million deaths annually worldwide (1). Approximately 70% of patients with HCC
are detected with unresectable or terminal stage cancer, leaving
only palliative treatment options such as TACE or sorafenib for
curative therapy (1,18).
The application of RT for treating HCC is limited by
critical hepatotoxicity (radiation-induced liver disease; RILD) at
doses lower than the therapeutic doses (19,20).
Recent advances in RT technology such as three-dimensional
conformal RT or stereotactic body RT enables the precise delivery
of a focused high drug dose on limited volumes of the tumorous
liver a allows reduction in the irradiation doses to the remaining
non-tumorous liver in order to minimize toxicity (21–24).
RT can be considered a salvage treatment option for inoperable HCC
that is unsuitable or refractory for TACE therapy, as well as a
potentially curative option for operable HCC that is unsuitable for
surgery or RFA (25–27). Despite the noteworthy development of
RT technology, effective RT is often unsatisfactory owing to
suboptimal delivery of doses associated with poor liver function
reserves or large tumor sizes. In such cases, the study of the
mechanism of radioresistance is important to optimize the
irradiation effect.
Irradiation activates transcription factors such as
NF-κB that can upregulate anti-apoptosis, pro-survival and invasive
signaling to confer radioresistance (7). PARP1 has been essential for
irradiation-induced NF-κB activation. Additionally, inhibition of
PARP1 increases cell death by irradiation and decreases the
X-linked inhibitor of apoptosis expression in breast cancer cell
lines (28). Overexpression of
cyclin D1 is associated with acquired radioresistance in HeLa (a
cervical cancer cell line) cells that was induced by fractionated
radiation. The inhibition of cyclin D1 by using small interfering
RNA (siRNA) decreased the radioresistance (29).
Hypoxia in tumors is associated with the induction
of radioresistance. HCC cells are frequently exposed to hypoxic
conditions during several mechanisms. Intrinsic tumor
characteristics of HCC are also associated with hypoxia. HCC
generally develops through chronic hepatitis or cirrhosis, which
can damage hepatic blood supply. Highly proliferative
characteristics of tumor cells induce local hypoxia inside HCC due
to a shortage of blood supply. The extrinsic modification by
anticancer treatment is also associated with inducing hypoxia in
cancer cells; for example, TACE induces hypoxia in tumor via
embolization of the tumor-feeding artery. Absorption of the
ionizing radiation by tissue leads to the generation of free
radicals and reactive oxygen species, which are chemically active
oxygen molecules that induce oxidative stress under which chemical
bonds break and a chain of events is initiated that results in DNA
damage. Oxygen molecules can react with these free radicals to
repair the DNA damage. Therefore, hypoxia interferes with the
repair of DNA damage caused by irradiation and induces
radioresistance (8,9).
Hypoxia also activates several hypoxia-induced
signaling factors such as HIF-1α, vascular endothelial growth
factor (VEGF), histone-modifying enzymes such as histone
deacetylase, and demethylase. High levels of serum VEGF are
associated with poor tumor response and the survival rates of
patients with advanced HCC who received TACE (30–33).
Findings of previous study showed that inhibition of HIF-1α by
siRNA decreased radioresistance in chemical hypoxic SMMC-7221
(human HCC cell line) cells (34).
Jumonji C-terminal-domain-containing histone demethylase
(JHDM) gene, JMJD1A, acts as a co-activator of
nuclear hormone receptors by demethylating dimethyl lysine 9 on
histone H3 (H3K9me2) of the target promoters. Findings of recent
study showed that the expression of JMJD1A mRNA is increased in
hypoxic PLC, HepG2, and Huh7 (human HCC cell lines) cells and that
the inhibition of JMJD1A by siRNA enhanced the cell-killing effect
(35). It has been suggested that
JMJD1A decreased H3K9 methylation and induced the target gene such
as adrenomedullin and that this cascade was regulated by HIF-1α
(36).
Emodin possesses anticancer, antibacterial, and
anti-inflammatory properties (37–40).
Emodin has been shown to inhibit cell growth by suppressing NF-κB,
increasing apoptosis and arresting the cell cycle at the G2/M phase
in human HCC cell lines via stimulation of p53 expression (11–13).
Emodin has also been shown to induce apoptosis in human cervical
cancer cells via the induction of PARP1 cleavage and caspase-9
activation (41). In addition,
emodin inhibited hypoxia-induced signaling factors such as HIF-1α,
VEGF, and histone deacetylase in several cell lines (10,14,15).
In this study, we have shown that hypoxia induced
radio-resistance in HepG2 cells and that the combination treatment
of emodin and radiation attenuated radioresistance. We also found
that emodin increased apoptosis and the G2/M phase arrest of HepG2.
We suggest that the possible mechanism of radioresistance
attenuation induced by hypoxia is a result of the upregulation of
apoptotic signaling factors such as cleaved PARP1, caspase-9, and
p53. These findings are associated with apoptosis and are
consistent with findings reported by previous studies (42–44).
Moreover, we have shown that epigenetic signaling such as JMJD2B
was maximally downregulated in the combination of emodin and
irradiation even though emodin only slightly inhibited
hypoxia-induced signaling factors such as HIF-1α, and histone
demethylase (JMJD1A). We therefore suggest that the emodin dose
used in this study was not sufficient to suppress HIF-1α, because
higher doses (25 and 50 μM) of emodin have been previously reported
to satisfactorily inhibit HIF-1α and VEGF (10,15).
Future studies using different doses of emodin are necessary to
investigate the epigenetic mechanism involved in hypoxic HCC.
The limitations of this study include that the
experiments were performed using only one human HCC cell line and
no in vivo experiments were conducted. In addition, although
emodin induced the additional suppression of HepG2 cells, it cannot
be concluded that emodin completely overcame the radioresistance of
hypoxic HCC. Future studies using a wide range of emodin doses are
required to completely understand the synergistic cell-killing
effect of emodin in hypoxic HCC.
In conclusion, emodin can attenuate radioresistance,
induced by hypoxia, in HepG2 cells via the enhancement of PARP1
cleavage, activation of caspase-9 and inhibition of JMJD2B. Thus,
our findings can provide new insights of the pharmacological
mechanism of emodin and its role as a radiosensitizer in HCC as
well as facilitate designing new therapeutic strategies for
radioresistant HCC.
Acknowledgements
This study was supported by the National R&D
Program through the Dongnam Institute of Radiological & Medical
Sciences (DIRAMS) funded by the Ministry of Education, Science, and
Technology (50595-2014).
References
|
1
|
Bruix J and Sherman M: Management of
hepatocellular carcinoma. Hepatology. 42:1208–1236. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee IJ and Seong J: Radiotherapeutic
strategies in the management of hepatocellular carcinoma. Oncology.
81(Suppl 1): 123–133. 2011. View Article : Google Scholar
|
|
3
|
Ursino S, Greco C, Cartei F, et al:
Radiotherapy and hepatocellular carcinoma: update and review of the
literature. Eur Rev Med Pharmacol Sci. 16:1599–1604.
2012.PubMed/NCBI
|
|
4
|
Feng M and Ben-Josef E: Radiation therapy
for hepatocellular carcinoma. Semin Radiat Oncol. 21:271–277. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kang JK, Kim MS, Cho CK, et al:
Stereotactic body radiation therapy for inoperable hepatocellular
carcinoma as a local salvage treatment after incomplete
transarterial chemoembolization. Cancer. 118:5424–5431. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meng MB, Cui YL, Lu Y, et al:
Transcatheter arterial chemo-embolization in combination with
radiotherapy for unresectable hepatocellular carcinoma: a
systematic review and meta-analysis. Radiother Oncol. 92:184–194.
2009. View Article : Google Scholar
|
|
7
|
Deorukhkar A and Krishnan S: Targeting
inflammatory pathways for tumor radiosensitization. Biochem
Pharmacol. 80:1904–1914. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stea B and Gordon J: Clinically relevant
biomarkers in targeted radiotherapy. Clin Exp Metastasis.
29:853–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meijer TW, Kaanders JH, Span PN and
Bussink J: Targeting hypoxia, HIF-1, and tumor glucose metabolism
to improve radiotherapy efficacy. Clin Cancer Res. 18:5585–5594.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu HF, Lai KC, Hsu SC, et al: Involvement
of matrix metallo-proteinases on the inhibition of cells invasion
and migration by emodin in human neuroblastoma SH-SY5Y cells.
Neurochem Res. 34:1575–1583. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei WT, Lin SZ, Liu DL and Wang ZH: The
distinct mechanisms of the antitumor activity of emodin in
different types of cancer (Review). Oncol Rep. 30:2555–2562.
2013.PubMed/NCBI
|
|
12
|
Hsu CM, Hsu YA, Tsai Y, et al: Emodin
inhibits the growth of hepatoma cells: finding the common
anti-cancer pathway using Huh7, Hep3B, and HepG2 cells. Biochem
Biophys Res Commun. 392:473–478. 2010. View Article : Google Scholar
|
|
13
|
Shieh DE, Chen YY, Yen MH, Chiang LC and
Lin CC: Emodin-induced apoptosis through p53-dependent pathway in
human hepatoma cells. Life Sci. 74:2279–2290. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Park SC, Yoon JH, Lee JH, et al:
Hypoxia-inducible adrenomedullin accelerates hepatocellular
carcinoma cell growth. Cancer Lett. 271:314–322. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang XZ, Wang J, Huang C, et al: Emodin
enhances cyto-toxicity of chemotherapeutic drugs in prostate cancer
cells: the mechanisms involve ROS-mediated suppression of multidrug
resistance and hypoxia inducible factor-1. Cancer Biol Ther.
7:468–475. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park SJ, Kim JG, Son TG, et al: The
histone demethylase JMJD1A regulates adrenomedullin-mediated cell
proliferation in hepatocellular carcinoma under hypoxia. Biochem
Biophys Res Commun. 434:722–727. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim JG, Yi JM, Park SJ, et al: Histone
demethylase JMJD2B-mediated cell proliferation regulated by hypoxia
and radiation in gastric cancer cell. Biochim Biophys Acta.
1819:1200–1207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schwarz RE and Smith DD: Trends in local
therapy for hepatocellular carcinoma and survival outcomes in the
US population. Am J Surg. 195:829–836. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cochrane AM, Murray-Lyon IM, Brinkley DM
and Williams R: Quadruple chemotherapy versus radiotherapy in
treatment of primary hepatocellular carcinoma. Cancer. 40:609–614.
1977. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ingold JA, Reed GB, Kaplan HS and Bagshaw
MA: Radiation Hepatitis. Am J Roentgenol Radium Ther Nucl Med.
93:200–208. 1965.PubMed/NCBI
|
|
21
|
Robertson JM, McGinn CJ, Walker S, et al:
A phase I trial of hepatic arterial bromodeoxyuridine and conformal
radiation therapy for patients with primary hepatobiliary cancers
or colorectal liver metastases. Int J Radiat Oncol Biol Phys.
39:1087–1092. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seong J, Keum KC, Han KH, et al: Combined
transcatheter arterial chemoembolization and local radiotherapy of
unresectable hepatocellular carcinoma. Int J Radiat Oncol Biol
Phys. 43:393–397. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shim SJ, Seong J, Han KH, Chon CY, Suh CO
and Lee JT: Local radiotherapy as a complement to incomplete
transcatheter arterial chemoembolization in locally advanced
hepatocellular carcinoma. Liver Int. 25:1189–1196. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park W, Lim DH, Paik SW, et al: Local
radiotherapy for patients with unresectable hepatocellular
carcinoma. Int J Radiat Oncol Biol Phys. 61:1143–1150. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shin YJ, Kim MS, Yoo SY, et al: Pilot
study of stereotactic body radiotherapy for huge hepatocellular
carcinoma unsuitable for other therapies. Tumori. 96:65–70.
2010.PubMed/NCBI
|
|
26
|
Seo YS, Kim MS, Yoo SY, et al: Preliminary
result of stereotactic body radiotherapy as a local salvage
treatment for inoperable hepatocellular carcinoma. J Surg Oncol.
102:209–214. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dawson LA: Overview: Where does radiation
therapy fit in the spectrum of liver cancer local-regional
therapies? Semin Radiat Oncol. 21:241–246. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Veuger SJ, Hunter JE and Durkacz BW:
Ionizingradiation-induced NF-kappaB activation requires PARP-1
function to confer radioresistance. Oncogene. 28:832–842. 2009.
View Article : Google Scholar :
|
|
29
|
Shimura T, Kakuda S, Ochiai Y, et al:
Acquired radioresistance of human tumor cells by
DNA-PK/AKT/GSK3beta-mediated cyclin D1 overexpression. Oncogene.
29:4826–4837. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chao Y, Li CP, Chau GY, et al: Prognostic
significance of vascular endothelial growth factor, basic
fibroblast growth factor, and angiogenin in patients with
resectable hepatocellular carcinoma after surgery. Ann Surg Oncol.
10:355–362. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jeng KS, Sheen IS, Wang YC, et al:
Prognostic significance of preoperative circulating vascular
endothelial growth factor messenger RNA expression in resectable
hepatocellular carcinoma: a prospective study. World J
Gastroenterol. 10:643–648. 2004.PubMed/NCBI
|
|
32
|
Poon RT, Ho JW, Tong CS, Lau C, Ng IO and
Fan ST: Prognostic significance of serum vascular endothelial
growth factor and endostatin in patients with hepatocellular
carcinoma. Br J Surg. 91:1354–1360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shim JH, Park JW, Kim JH, et al:
Association between increment of serum VEGF level and prognosis
after transcatheter arterial chemoembolization in hepatocellular
carcinoma patients. Cancer Sci. 99:2037–2044. 2008.PubMed/NCBI
|
|
34
|
Yang W, Sun T, Cao J and Fan S:
Hypoxia-inducible factor-1α downregulation by small interfering RNA
inhibits proliferation, induces apoptosis, and enhances
radiosensitivity in chemical hypoxic human hepatoma SMMC-7721
cells. Cancer Biother Radiopharm. 26:565–571. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yamada D, Kobayashi S, Yamamoto H, et al:
Role of the hypoxia-related gene, JMJD1A, in hepatocellular
carcinoma: clinical impact on recurrence after hepatic resection.
Ann Surg Oncol. 19(Suppl 3): 355–364. 2012. View Article : Google Scholar
|
|
36
|
Krieg AJ, Rankin EB, Chan D, Razorenova O,
Fernandez S and Giaccia AJ: Regulation of the histone demethylase
JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene
expression and tumor growth. Mol Cell Biol. 30:344–353. 2010.
View Article : Google Scholar
|
|
37
|
Andersen DO, Weber ND, Wood SG, Hughes BG,
Murray BK and North JA: In vitro virucidal activity of selected
anthraquinones and anthraquinone derivatives. Antiviral Res.
16:185–196. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Arosio B, Gagliano N, Fusaro LM, et al:
Aloe-emodin quinone pretreatment reduces acute liver injury induced
by carbon tetra-chloride. Pharmacol Toxicol. 87:229–233. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Krumbiegel G and Schulz HU: Rhein and
aloe-emodin kinetics from senna laxatives in man. Pharmacology.
47(Suppl 1): 120–124. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Srinivas G, Babykutty S, Sathiadevan PP
and Srinivas P: Molecular mechanism of emodin action: transition
from laxative ingredient to an antitumor agent. Med Res Rev.
27:591–608. 2007. View Article : Google Scholar
|
|
41
|
Srinivas G, Anto RJ, Srinivas P,
Vidhyalakshmi S, Senan VP and Karunagaran D: Emodin induces
apoptosis of human cervical cancer cells through poly(ADP-ribose)
polymerase cleavage and activation of caspase-9. Eur J Pharmacol.
473:117–125. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee KB, Kim KR, Huh TL and Lee YM: Proton
induces apoptosis of hypoxic tumor cells by the p53-dependent and
p38/JNK MAPK signaling pathways. Int J Oncol. 33:1247–1256.
2008.PubMed/NCBI
|
|
43
|
Hennessey D, Martin LM, Atzberger A, Lynch
TH, Hollywood D and Marignol L: Exposure to hypoxia following
irradiation increases radioresistance in prostate cancer cells.
Urol Oncol. 31:1106–1116. 2013. View Article : Google Scholar
|
|
44
|
Wang M, Li X, Qu Y, Xu O and Sun Q:
Hypoxia promotes radio-resistance of CD133-positive Hep-2 human
laryngeal squamous carcinoma cells in vitro. Int J Oncol.
43:131–140. 2013.PubMed/NCBI
|