Introduction
The human telomerase complex consists of an RNA
component (hTR), which acts as a template for the telomeric repeat
synthesis and human telomerase reverse transcriptase (hTERT)
(1). Telomerase activity is closely
correlated with cell proliferation and is required for the
unlimited proliferation potential of cancer cells (2). Active telomerase is found in ~80% of
cancer cells and rapidly proliferating tissues during early
development (3). Increased hTERT
expression leads to enhanced telomerase activity, which in turn,
affects cell ageing, cell cycle progression and tumorigenesis
(4). As such, targeting hTERT
represents a promising strategy for the development of anticancer
drugs.
Reflux of duodenal contents has been shown to be
involved in the pathogenesis of Barrett’s esophagus (5,6).
Moreover, bile acid was found to promote intestinal metaplasia and
gastric carcinogenesis (7). For
example, DCA and CDCA, the main constituents of the refluxate (pH
4.0–7.0) (8), have been reported to
promote esophageal carcinogenesis likely by activating the tuberous
sclerosis complex 1/mammalian target of rapamycin (TSC1/mTOR) or
nuclear factor κB (NF-κB) pathways (9,10).
Bile acids under acidified media were recently reported to
upregulate the expression of the proto-oncogene c-myc in Barrett’s
metaplasia and esophageal adenocarcinoma (11). c-myc is a gene transcription factor
and upregulated in many types of cancer. Among the well-documented
direct targets of c-myc is hTERT (12). Expression of c-myc in human
epithelial cells and fibroblasts induces the transcription of
endogenous hTERT; thereby activating telomerase (4). Hence, we hypothesized that acidified
bile acids may upregulate hTERT gene transcription by activating
c-myc in gastric cancer cells.
In the present study, we investigated the effects of
acidified bile acids (pH 5.5) on hTERT gene expression and
telomerase activity in gastric adenocarcinoma cells and explored
the underlying molecular mechanisms. We found that acidified bile
acids enhanced the expression and activity of hTERT by increasing
c-myc levels in gastric adenocarcinoma cells.
Materials and methods
Reagents
DCA, CDCA and small-molecular-weight c-myc inhibitor
(10058-F4; [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one)
were obtained from Sigma (St. Louis, MO, USA) and dissolved in
dimethylsulfoxide (DMSO). Rabbit monoclonal antibodies specific for
hTERT and c-myc were obtained from Sigma. TurboFect™ Transfection
reagent was acquired from Thermo Fisher. Dual-Glo®
Luciferase Assay system was from Promega. RNeasy total RNA
extraction kit was from Qiagen (Hilden, Germany). Takara Taq
DNA polymerase was from Takara Bio (Dalian, China).
Cell culture and treatment
Human gastric cancer cell lines MKN28, MGC803 and
SGC7901 were maintained in RPMI-1640 medium supplemented with 10%
calf fetal serum, 100 U/ml penicillin and 0.1 mg/ml streptomycin.
The three cell lines were treated as follows: i) acidified growth
media at pH 5.5 only; ii) acidified growth media at pH 5.5
containing 100 μM DCA; iii) growth media at pH 7.0
containing 100 μM DCA; iv) acidified growth media at pH 5.5
containing 100 μM CDCA; and v) growth media at pH 7.0
containing 100 μM CDCA. Cells were stimulated continuously for 12
or 24 h and then incubated in neutral growth media (growth media at
pH 7.0) for 3 h. For inhibitor assays, cells were pretreated with
inhibitors for 24 h before exposure to acidified growth media
containing bile acids. All experiments were performed in
triplicate.
RNA isolation and reverse
transcription-polymerase chain reaction (RT-PCR)
Cells were harvested after treatment and total RNA
was extracted with an RNA extraction kit from Qiagen. Total RNA was
first transcribed into cDNA in 25 μl of reagent mix, 1
μl of which was amplified by PCR in a 25 μl reaction
using Takara Taq DNA polymerase with 0.5 μl primers
(20 μM). Primers for hTERT were: (forward)
5′-ATCAGACAGCACTTGAAGAG-3′ and (reverse)
5′-GTAGTCCATGTTCACAATCG-3′; for c-myc, (forward)
5′-TCAAGAGGTGCCACGTCTCC-3′ and (reverse)
5′-TCTTGGCAGCAGGATAGTCCTT-3′. The PCR reaction mixture was
denatured at 94°C for 3 min followed by 30 cycles at 94°C for 30
sec, 51°C (TERT) and 61°C (c-myc) for 30 sec, and 72°C for 60 sec.
Primers for β-actin, (forward) 5′-CTTAGCACCCCTGGCCAAG-3′, (reverse)
5′-GATGTTCTGGAGAGCCCCG-3′ were used to amplify β-actin as an
internal control. Primers were synthesized by Sangon Biotech
(Shanghai, China). PCR products were analyzed by Image Lab version
4.1 software.
Western blotting
Cells were lysed on ice with NP-40 buffer containing
40 mM Tris-HCI, pH 6.9, 2 mM ethylenediaminetetraacetic acid, 100
mM sodium fluoride, 150 mM NaCl, 10 mM sodium pyrophosphate, 1%
Nonidet P40 (NP-40), 2 mM orthovanadate, 1% Triton X-100, 0.3 mM
phenylmethanesulfonyl fluoride, and 1 Mini Tablet Protein Inhibitor
(Roche, South San Francisco, CA, USA). Cell lysates were
centrifuged at 12,000 × g for 10 min at 4°C, and the supernatants
were stored at −80°C. Protein quantification was performed with the
BCA protein assay (Pierce, Rockford, IL, USA). Equal amounts of
protein from different groups were denatured in SDS sample buffer
and separated on 10% sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred onto polyvinylidene
difluoride membranes. β-actin, hTERT and c-myc antibodies (1:2,000
dilution) were used to detect the corresponding proteins. Blots
were developed using SuperSignal West Femto Maximum Sensitivity
Substrate (Thermo Scientific Pierce, Rockford, IL, USA).
Transient transfection and luciferase
reporter assays
Plasmids were prepared using the Omega Plasmid Midi
kit from Omega Bio-Tek Inc. (Norcross, GA, USA). The 295-bp
promoter (13) region of the human
TERT gene (GenBank accession no. AN097365) was amplified by PCR
from human genomic DNA, using the following primer pairs: TERT
(forward) 5′-CTAGCTAGCCACAGACGCCCAGGACCGCGCTTC-3′ and (reverse)
5′-CCCAAGCTTCCACGTGCGCCCACGTGCGCCCAC-3′. The 295-bp fragment was
inserted upstream of the luciferase reporter pGL4.20 vector
(Promega) and verified by DNA sequencing to obtain plasmid
pgl4.20-hTERTp.
The 225-bp promoter (14,15) of
the human v-myc avian myelocytomatosis viral oncogene c-myc gene
(GenBank accession no. NG007161) was generated by PCR from human
genomic DNA, using the following primer pairs: (forward)
5′-ATAGATCTCTCTTACTCTGTTTACATCCTAGAGC-3′
and (reverse) 5′-CTAAGCTTCCGGGAGGGGCGCTTATGGGGAGGG-3′.
The 225-bp amplified fragment was cloned into pGL4.20 to obtain the
plasmid pGL4.20-mycp (15).
The cells were seeded at a concentration of 5,000
cells/well in 96-well plates the day before transfection. The cells
were transfected with plasmids pGL4.20-hTERTp or pGL4.20-mycp
together with pGL4.74 containing the TK promoter using TurboFect™
Transfection reagent following the manufacturer’s protocol.
Twenty-four hours later, the cells were incubated in different
media with or without bile acids or inhibitors for an additional 24
h, followed by measurement of the luciferase activity.
Telomerase activity assay
Telomerase activity was assayed by the stretch PCR
method (16–18) using the TeloChaser (Toyobo Co.,
Ltd., Osaka, Japan) according to the manufacturer’s instructions.
In brief, cells were pelleted and washed in ice-cold PBS buffer.
After washing, the pellets were homogenized in a 200 μl
volume of ice-cold lysis buffer and incubated on ice for 30 min.
The lysate was centrifuged at 15,000 rpm for 20 min at 4°C. The
supernatant was frozen and stored at −80°C. Nine micrograms of
protein were used for the PCR assay. For the negative control, each
extract was pre-incubated with 1 μg of RNase A for 30 min at
37°C. After incubation at 37°C for 60 min, the DNA products were
isolated, heated at 90°C for 3 min and subjected to 30 cycles of
PCR including denaturation at 94°C for 45 sec, annealing at 50°C
for 30 sec, and extension at 72°C for 60 sec. The PCR products were
electrophoresed on a 10% polyacrylamide gel and stained with
ethidium bromide. Images were captured using the FLA-3000g image
analyzer (Fuji Film Corp., Tokyo, Japan).
Statistical analyses
Results are shown as means ± standard error.
Differences were evaluated with unpaired two-tailed Student’s
t-tests with unequal variance for multiple comparisons using the
SPSS software, version 16.0. The difference between two values was
considered statistically significant at P<0.05. Experiments were
repeated independently at least three times.
Results
Bile acids in acidified growth media
upregulate hTERT expression at the transcriptional level in gastric
cancer cells
To determine the effect of bile acids in acidified
growth media on hTERT expression in gastric cancer cells, we used
the bile acids DCA and CDCA as they are common constituents of the
refluxate and are widely studied in cell culture models. A
concentration of 100 μM was used as it has been shown to be
a physiological concentration in our cell culture model (11). In addition, as the pH of reflux
constituents ranges between 4.0 and 7.0 (8), we used a median value pH value of 5.5.
We treated human gastric cancer MKN28, MGC803 and SGC7901 cells
with 100 μM DCA or CDCA with acidified growth media at pH
5.5 for 12 or 24 h and determined the mRNA and protein levels of
hTERT by RT-PCR and western blotting, respectively. In the three
cell lines, the addition of 100 μM of either DCA or CDCA to
acidified growth media at pH 5.5 for either 12 or 24 h
significantly increased hTERT mRNA expression (Fig. 1). In sharp contrast, the mRNA levels
of hTERT were not affected by DCA or CDCA at pH 7.0, or acidified
media alone over 24 h (Fig. 2A).
Consistent with the induction of hTERT mRNA, western blotting
results showed that either DCA or CDCA at pH 5.5 but not at pH 7.0
significantly increased the protein levels of hTERT in all the 3
cell lines (Fig. 3). These results
demonstrated that DCA and CDCA under acidic conditions upregulated
the expression of hTERT in gastric cancer cells.
To determine whether bile acids increase hTERT
promoter activity, we employed an hTERT promoter luciferase
construct pGL4.20-hTERTp, which contains a 295-bp human hTERT
promoter core region fused to a luciferase reporter gene. After
transient transfection of human gastric cancer cells with
pGL4.20-hTERTp for 24 h, the cells were treated with different
conditions and luciferase activities were determined. As shown in
Fig. 4A, 100 μM DCA or CDCA
with media of pH 5.5 induced the hTERT promoter driven luciferase
activities in these cells, with a maximum 3-fold increase with CDCA
in the SgC7901 cells. These results indicated that acidified bile
acids in acidic conditions but not neutral pH induced hTERT
upregulation at the transcriptional level.
Bile acids in acidified growth media
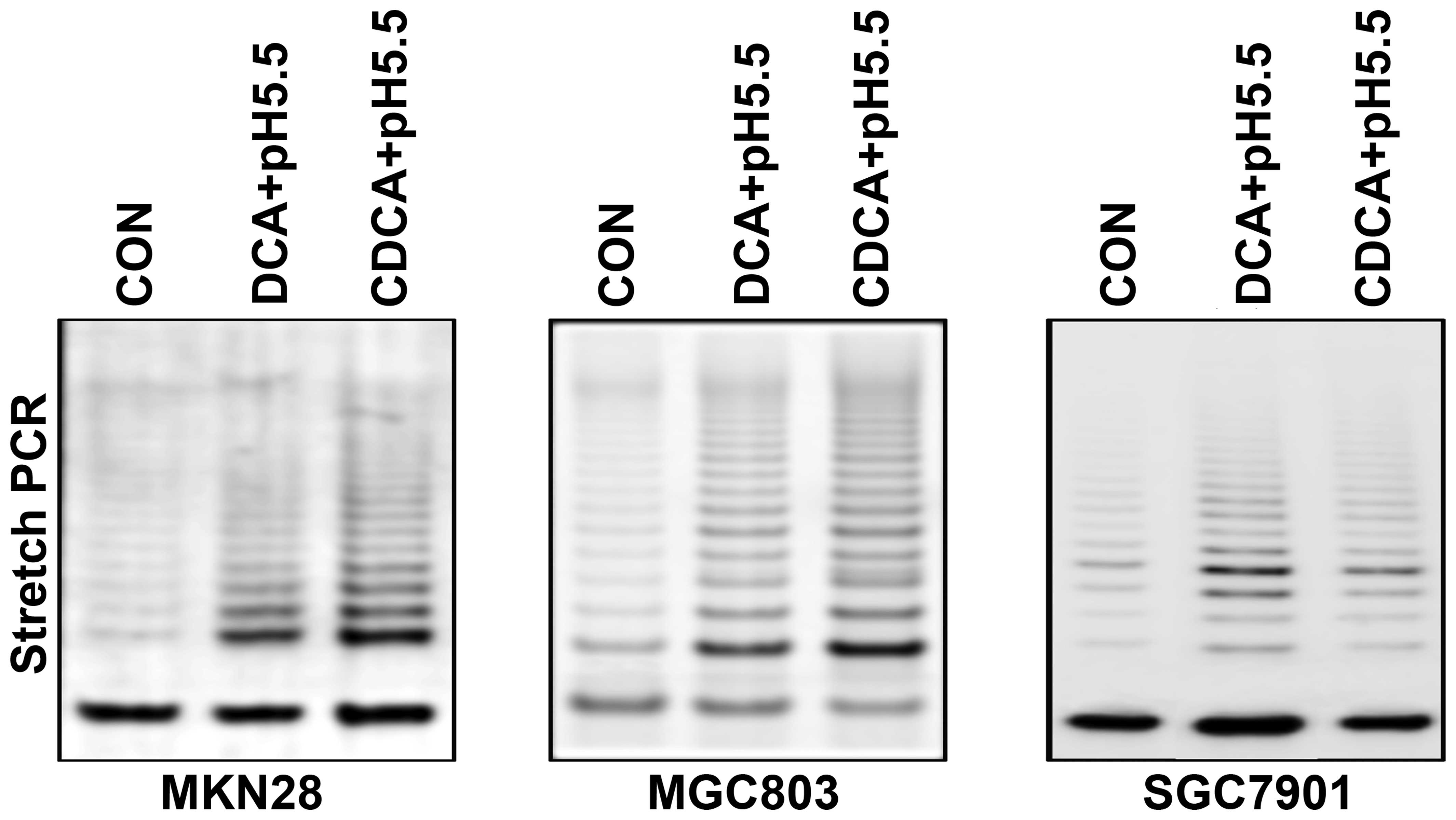
increase the telomerase activity in gastric cancer cells
We assessed the telomerase activity of gastric
cancer cells stimulated with bile acids in acidic conditions.
Specifically, three gastric cancer cell lines were treated with 100
μM DCA or CDCA with acidified media for 24 h. Telomerase
activity was then measured by stretch PCR and expressed as a ladder
of 6-bp bands or multiples of 6-bp intervals. The percentage of
telomerase activity was calculated using the band intensity.
Telomerase activity was significantly increased following treatment
with bile acids in acidified growth media when compared with the
untreated cells (Fig. 5).
Bile acids in acidified growth media
induce c-myc expression at the transcriptional level in gastric
cancer cells
Previous reports have suggested that c-myc is
important in mediating bile acid signaling (11). To test whether bile acids in
acidified growth media induce the expression of hTERT via c-myc, we
evaluated c-myc protein levels and gene transcription in 3 human
gastric cancer cell lines. Indeed, in all the three cell lines, DCA
or CDCA with acidified media increased c-myc mRNA expression in a
time-dependent fashion with a maximum increase of ~7-fold (Fig. 1). Moreover, consistent with changes
in hTERT mRNA levels, DCA or CDCA at pH 7.0, or acidified media
alone did not increase c-myc gene transcription. In contrast, DCA
or CDCA under acidified media conditions elicited clear effects
(Fig. 2A). To determine whether
bile acids increase c-myc promoter activity, we employed a c-myc
promoter luciferase construct pGL4.20-mycp containing a 225-bp
human c-myc promoter core region. We found that DCA and CDCA with
acidified media induced c-myc promoter driven luciferase activities
in these cells, with a maximum increase of 7.5-fold using 100
μM DCA in MKN28 cells. In contrast, 100 μM DCA or
CDCA at pH 7.0, or acidified media alone failed to induce c-myc
promoter driven luciferase activity at 24 h (Fig. 4B). These results indicated that
acidic conditions were indeed required for bile acid to induce
c-myc expression at the transcriptional level.
Pharmacologic inhibition of c-myc
prevents the induction of hTERT by DCA or CDCA under acidic
conditions in gastric cancer cells
As hTERT is a direct target of c-myc, our results
suggested that induction of c-myc is one of the mechanisms by which
bile acids under acidified growth media increase hTERT expression
in gastric cancer cells. To test this hypothesis, we pretreated
these cells with 10058-F4, a specific inhibitor of c-myc, followed
by incubation with bile acids in acidified growth media. The
protein levels and gene transcription of c-myc and hTERT were
determined by immunoblotting and RT-PCR, respectively. In all three
cell lines, 10058-F4 reduced the mRNA levels of c-myc and hTERT
compared to the control. Intriguingly, pretreatment with 100
μM 10058-F4 for 24 h (19,20)
significantly abolished the induction of the mRNA levels of c-myc
and hTERT by either DCA or CDCA in media at pH 5.5 (Fig. 2B). Consistent with the effect on
c-myc and hTERT mRNA levels, western blot analysis demonstrated
that either DCA or CDCA at pH 5.5 significantly prevented the
induction of the protein levels of c-myc and hTERT in all 3 cell
lines (Fig. 3). These results
implied that DCA and CDCA under acidic conditions upregulated the
expression of hTERT by activating c-myc in gastric cancer
cells.
To extend our observations further, we asked whether
10058-F4 blocked hTERT promoter activity. We transiently
transfected cells with plasmid pGL4.20-hTERTp or pGL4.20-mycp,
treated with 100 μM DCA or CDCA under acidified growth media
for 24 h with or without 10058-F4, and then determined the
luciferase activities. In the 3 human gastric cancer cell lines,
10058-F4 inhibited hTERT and c-myc promoter-driven luciferase
activities and apparently prevented the induction of hTERT
(Fig. 4A) and c-myc (Fig. 4B) promoter-driven luciferase
activities by DCA or CDCA under acidified growth media. Our studies
indicated that pharmacologic inhibition of c-myc prevented the
induction of hTERT by both DCA and CDCA in acidic conditions in
gastric adenocarcinoma cells, suggesting that activation of c-myc
contributes to the induction of hTERT by bile acids under acidified
growth media in gastric cancer cells.
Discussion
In the present study, we found that bile acids in
acidified media significantly increased the expression of both
c-myc and hTERT in gastric cancer cells. This increase was
abolished by the c-myc specific inhibitor 10058-F4. Our results
demonstrated that bile acids under acidified conditions induced
hTERT overexpression in human gastric cancer cells by activation of
c-myc transcription and suggest that acidified bile acids may
promote tumorigenesis via activation of telomerase.
Reflux of juices from the duodenum has been closely
associated with the development of gastric adenocarcinoma (21–23),
but the effects of duodenal gastric reflux (DGR) on the
proliferation of gastric cancer have not yet been well studied. The
unconjugated bile acids DCA and CDCA have been shown to be the main
components in DGR (24), and have
been widely studied in cell culture models. Moreover, bile acids
under acidified media have been reported to induce the c-myc gene
expression in esophageal cells, but the molecular events
responsible for hTERT gene expression and telomerase activity have
not been studied in gastric adenocarcinoma cells (11). In the present study, we found that
bile acids increased hTERT expression at the transcriptional level
in gastric cancer cells only under acidic conditions. Furthermore,
this effect depended on c-myc expression.
The telomerase RNA subunit is essential for
telomerase activity, and is expressed in both normal cells and
cells that continuously divide beyond replicative senescence
(25). Data from both in
vitro and in vivo studies clearly demonstrate that TERT
is the determinant of telomerase activity (26). The transcriptional activity of the
TERT gene promoter is significantly higher in telomerase-positive
cells than in telomerase-negative cells (2). In addition, studies with
dual-luciferase gene assays indicated that hTERT gene expression is
controlled mainly at the transcriptional rather than the
post-transcriptional level. As telomere length is thought to be the
limiting factor in determining the lifespan of a cell (27), stimulation of telomerase activity
may be important for the ability of hTERT to promote cell
proliferation. We found that exposure of three gastric
adenocarcinoma cell lines to acidified bile acids induced the
expression of hTERT, an effect that was accompanied by increased
telomerase activity. Our data suggest that acidified bile acids may
promote carcinogenesis of gastric cancer by stimulating the
expression of hTERT.
c-myc is an important transcription factor that
mediates expression of multiple genes in important biological
processes, including cell proliferation, growth arrest and
apoptosis (28). c-myc was recently
reported to enhance the activity of the hTERT promoter, an effect
that was completely abrogated by deletion of a single Myc/Max
binding site within the core promoter (12). However, Wu et al reported
that the 5′ region of human TERT contains 29 potential Myc/Max
binding sites, including 18 canonical (CACGTG) and 11 specific
non-canonical (CA(C/T)GCG) sites (29). Nonetheless, it was found that the
upregulation of TERT expression by c-myc was a direct effect and
was independent of new protein synthesis (29). Other transcription factors including
AP-1, c-Jun and Jun have been shown to regulate hTERT expression in
other contexts (30). Activity of
these transcription factors may also be required for hTERT
expression in gastric adenocarcinoma cells. In agreement with the
previous finding that c-myc regulates the expression of hTERT
(4), our data revealed that
10058-F4, an inhibitor of c-myc, reduced the endogenous as well as
acidified bile acid-mediated upregulation of hTERT. In addition,
c-myc expression and transcription activity coincided with hTERT
induction, while inhibition of c-myc markedly suppressed the
upregulation of hTERT by acidified bile acids, indicating that
c-myc is indeed involved in hTERT transcription induced by bile
acids under acidified media.
In conclusion, the present study revealed that
acidified bile acids induced c-myc and hTERT expression in gastric
adenocarcinoma cells. Our findings suggest that bile acid-induced
upregulation of c-myc may be a mechanism through which acidified
bile acids regulate telomerase activity in gastric cancer.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81172362 and
81172359), and the Co-ordinative and Innovative Plan Projects of
the Science and Technology in Shaanxi Province (2013KTCQ03-08). We
thank Medjaden Bioscience Ltd. for assisting in the preparation of
this manuscript.
References
|
1
|
Feng J, Funk WD, Wang SS, Weinrich SL,
Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC, Yu J, et al:
The RNA component of human telomerase. Science. 269:1236–1241.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Greider CW: Telomerase activity, cell
proliferation, and cancer. Proc Natl Acad Sci USA. 95:90–92. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
George J, Banik NL and Ray SK: Knockdown
of hTERT and concurrent treatment with interferon-gamma inhibited
proliferation and invasion of human glioblastoma cell lines. Int J
Biochem Cell Biol. 42:1164–1173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cerni C: Telomeres, telomerase, and myc.
An update Mutat Res. 462:31–47. 2000. View Article : Google Scholar
|
|
5
|
Zhang F, Altorki NK, Wu Y, Soslow RA,
Subbaramaiah K and Dannenberg AJ: Duodenal reflux induces
cyclooxygenase-2 in the esophageal mucosa of rats: evidence for
involvement of bile acids. Gastroenterology. 121:1391–1399. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goldman A, Chen HD, Roesly HB, Hill KA,
Tome ME, Dvorak B, Bernstein H and Dvorak K: Characterization of
squamous esophageal cells resistant to bile acids at acidic pH:
implication for Barrett’s esophagus pathogenesis. Am J Physiol
Gastrointest Liver Physiol. 300:G292–G302. 2011. View Article : Google Scholar
|
|
7
|
Tatsugami M, Ito M, Tanaka S, Yoshihara M,
Matsui H, Haruma K and Chayama K: Bile acid promotes intestinal
metaplasia and gastric carcinogenesis. Cancer Epidemiol Biomarkers
Prev. 21:2101–2107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kauer WK, Peters JH, DeMeester TR, Ireland
AP, Bremner CG and Hagen JA: Mixed reflux of gastric and duodenal
juices is more harmful to the esophagus than gastric juice alone.
The need for surgical therapy re-emphasized. Ann Surg. 222:525–533.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yen CJ, Izzo JG, Lee DF, Guha S, Wei Y, Wu
TT, Chen CT, Kuo HP, Hsu JM and Sun HL: Bile acid exposure
up-regulates tuberous sclerosis complex 1/mammalian target of
rapamycin pathway in Barrett’s-associated esophageal
adenocarcinoma. Cancer Res. 68:2632–2640. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jenkins GJ, Cronin J, Alhamdani A, Rawat
N, D’Souza F, Thomas T, Eltahir Z, Griffths AP and Baxter JN: The
bile acid deoxycholic acid has a non-linear dose response for DNA
damage and possibly NF-κB activation in oesophageal cells, with a
mechanism of action involving ROS. Mutagenesis. 23:399–405. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tselepis C, Morris CD, Wakelin D, Hardy R,
Perry I, Luong QT, Harper E, Harrison R, Attwood SE and Jankowski
JA: Upregulation of the oncogene c-myc in Barrett’s adenocarcinoma:
induction of c-myc by acidified bile acid in vitro. Gut.
52:174–180. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Greenberg RA, O’Hagan RC, Deng H, Xiao Q,
Hann SR, Adams RR, Lichtsteiner S, Chin L, Morin GB and DePinho RA:
Telomerase reverse transcriptase gene is a direct target of c-Myc
but is not functionally equivalent in cellular transformation.
Oncogene. 18:1219–1226. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Horikawa I, Chiang YJ, Patterson T,
Feigenbaum L, Leem SH, Michishita E, Larionov V, Hodes RJ and
Barrett JC: Differential cis-regulation of human versus mouse TERT
gene expression in vivo: identification of a human-specific
repressive element. Proc Natl Acad Sci USA. 102:18437–18442. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang C, Mayer JA, Mazumdar A, Fertuck K,
Kim H, Brown M and Brown PH: Estrogen induces c-myc gene expression
via an upstream enhancer activated by the estrogen receptor and the
AP-1 transcription factor. Mol Endocrinol. 25:1527–1538. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hoshi S, Takahashi T, Satoh M, Numahata K,
Suzuki K, Ohyama C, Mori M, Mituoka T, Nakagawara K and Orikasa S:
Telomerase activity. Urol Oncol. 5:25–30. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tomizawa A, Kanno SI, Osanai Y, Yomogida S
and Ishikawa M: Cytotoxic effects of caffeic acid undecyl ester are
involved in the inhibition of telomerase activity in NAlM-6 human B
cell leukemia cells. Oncol Lett. 6:875–877. 2013.PubMed/NCBI
|
|
18
|
Hasegawa T, Chosa N, Asakawa T, Yoshimura
Y, Ishisaki A and Tanaka M: Establishment of immortalized human
periodontal ligament cells derived from deciduous teeth. Int J Mol
Med. 26:701–705. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin CP, Liu JD, Chow JM, Liu CR and Liu
HE: Small-molecule c-Myc inhibitor, 10058-F4, inhibits
proliferation, downregulates human telomerase reverse transcriptase
and enhances chemosensitivity in human hepatocellular carcinoma
cells. Anticancer Drugs. 18:161–170. 2007. View Article : Google Scholar
|
|
20
|
Huang M, Cheng Y, Liu C, Lin S and Liu HE:
A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle
arrest, apoptosis, and myeloid differentiation of human acute
myeloid leukemia. Exp Hematol. 34:1480–1489. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Debruyne PR, Bruyneel EA, Li X, Zimber A,
Gespach C and Mareel MM: The role of bile acids in carcinogenesis.
Mutat Res. 480–481:359–369. 2001. View Article : Google Scholar
|
|
22
|
Bajpai M, Kessel R, Bhagat T, Nischal S,
Yu Y, Verma A and Das KM: High resolution integrative analysis
reveals widespread genetic and epigenetic changes after chronic in
vitro acid and bile exposure in Barretts epithelium cells. Genes
Chromosomes Cancer. 52:1123–1132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miwa K, Hasegawa H, Fujimura T, Matsumoto
H, Miyata R, Kosaka T, Miyazaki I and Hattori T: Duodenal reflux
through the pylorus induces gastric adenocarcinoma in the rat.
Carcinogenesis. 13:2313–2316. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dixon MF, Mapstone NP, Neville PM,
Moayyedi P and Axon AT: Bile reflux gastritis and intestinal
metaplasia at the cardia. Gut. 51:351–355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakayama J, Tahara H, Tahara E, Saito M,
Ito K, Nakamura H, Nakanishi T, Tahara E, Ide T and Ishikawa F:
Telomerase activation by hTRT in human normal fibroblasts and
hepatocellular carcinomas. Nat Genet. 18:65–68. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li C, Hsiao Y, Wu T, Lin Y, Yeh K and Ko
J: Vorinostat, SAHA, represses telomerase activity via epigenetic
regulation of telomerase reverse transcriptase in non-small cell
lung cancer cells. J Cell Biochem. 112:3044–3053. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bodnar AG, Ouellette M, Frolkis M, Holt
SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S and
Wright WE: Extension of life-span by introduction of telomerase
into normal human cells. Science. 279:349–352. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pelengaris S, Rudolph B and Littlewood T:
Action of Myc in vivo - proliferation and apoptosis. Curr Opin
Genet Dev. 10:100–105. 2000. View Article : Google Scholar
|
|
29
|
Wu KJ, Grandori C, Amacker M, Simon-Vermot
N, Polack A, Lingner J and Dalla-Favera R: Direct activation of
TERT transcription by c-MYC. Nat Genet. 21:220–224. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takakura M, Kyo S, Inoue M, Wright WE and
Shay JW: Function of AP-1 in transcription of the telomerase
reverse transcriptase gene (TERT) in human and mouse cells. Mol
Cell Biol. 25:8037–8043. 2005. View Article : Google Scholar : PubMed/NCBI
|