Introduction
Breast cancer is the most common cancer among women,
with an estimated 1.7 million new cases worldwide in 2012
[International Agency for Research on Cancer (IARC), 2012]. Tumor
invasion and metastasis affect more than 90% of patients with
breast cancer and are the main factors that lead to high mortality
(1). To date, there has been no
significant breakthrough in the control of breast cancer.
Epithelial-to-mesenchymal transition (EMT) is
believed to be one of the main factors that promotes breast cancer
progression. EMT has been known to impart metastasis and stemness
characteristics in several types of cancers, including breast
cancer, through inducing cancer cells to change from an epithelial
to a mesenchymal phenotype, resulting in cancer cell invasion and
metastasis (2–4). Furthermore, EMT also enhanced
multidrug resistance by upregulating ATP binding cassette
transporters in invasive breast cancer cells (5). The modulation of EMT includes numerous
gene regulatory networks, including microRNAs (miRNAs) (6).
miRNAs are small non-coding RNA molecules that
inhibit gene expression by interacting preferentially with the
3′-untranslated regions (3′-UTRs) of target mRNAs (7). Recently, a number of miRNAs, such as
miR-205, miR-21, miR-30a, miR-96 and miR-107 have been shown to be
involved in metastasis and cell proliferation, as well as EMT in
breast cancer (8,9). In particular, miR-494 appears to play
different roles in various types of tumors. miR-494 was shown to
induce tumor necrosis factor-related apoptosis-inducing ligand
(TRAIL) resistance in non-small cell lung cancer, and is considered
an an oncogenic miRNA mega cluster that regulates the G1/S
transition during liver tumorigenesis (10,11).
In contrast, miR-494 is downregulated in human cholangiocarcinomas,
prostate and breast cancer, and gastric carcinoma, and its
reinforcement results in cancer growth inhibition, which indicates
that miR-494 acts as an anti-oncogene (12). However, the role and the underlying
mechanism of miR-494 in breast cancer are still not fully
understood. In the present study, we focused on the role of miR-494
in the progression of breast cancer and the related molecular
mechanism.
Materials and methods
Cell lines, culture and treatment
The human breast cancer cell lines MDA-MB-231,
MCF-7, MDA-MB-468, MDA-MB-435, T47D, BT-474, SK-BR-3, ZR-75-30 and
human normal breast epithelial cell line MCF-10A were all obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). The 293T cells were also purchased from ATCC. The breast
cancer cells were cultured in Dulbecco’s modified Eagle’s medium
(DMEM) supplemented with 10% fetal bovine serum (both from Gibco,
Carlsbad, CA, USA), penicillin (100 U/ml) and streptomycin (100
μg/ml) (Enpromise, China). MCF-10A cells were cultured in
Mammary Epithelial Basal Medium (Lonza, Walkersville, MD, USA).
Cells were incubated at 37°C in a humidified chamber supplemented
with 5% CO2.
Upregulation and downregulation of
miR-494
miR-494 (mature sequence, UGAAACAUACACGGGAAACCUC)
mimic, inhibitor, miRNA mimic negative control and miRNA inhibitor
negative control were purchased from Sigma-Aldrich Inc. (St. Louis,
MO, USA). The MDA-MB-231 cells were cultured to ~30–40% confluency
in 6-well plates, and were transfected with miR-494 mimics, miRNA
mimic negative control, miR-494 inhibitors or miRNA inhibitor
negative control using Lipofectamine 2000 (Invitrogen, Carlsbad,
CA, USA), in accordance with the manufacturer’s instructions. After
48 h of incubation, cells were harvested for further analysis. All
transfections were performed in triplicate.
CXCR4 siRNA transfection
For targeted knockdown of chemokine (C-X-C motif)
receptor 4 (CXCR4), two siRNAs targeted to CXCR4 were designed
according to a previous study (13)
and synthesized by Genetimes Technology (Shanghai, China). The
sequence were: 5′-UAAAAUCUUCCUGCCCCCdTdT-3′ and
5′-GGAAGCUGUUGGCUGAAAAdTdT-3′. The non-specific control siRNA was
provided by Genetimes Technology. For transfection,
5×104 MDA-MB-231 cells seeded in each cell of 24-well
microplates were transfected with siRNAs at a final concentration
of 100 nmol/l using Lipofectamine 2000.
Quantitative reverse
transcrption-polymerase chain reaction
Total RNA from cultured cells was isolated using
TRIzol reagent (Life Technologies, Inc., Rockville, MD, USA)
according to the manufacturer’s instructions. Reverse transcription
was performed on 1 μg of total RNA from each sample using
oligo(dT) primers and 200 units of SuperScript II (Life
Technologies, Inc.) for extension. Quantitative real-time RT-PCR
(qRT-PCR) analysis of CXCR4, β-catenin, lymphoid enhancer-binding
factor 1 (LEF1), cyclin-D1, CD44, E-cadherin, N-cadherin, vimentin,
α-smooth muscle actin (α-SMA) and β-actin was carried out using the
primers shown in Table I with the
SYBR-Green Master Mix (Takara, Dalian, China) according to the
manufacturer’s instructions. PCR amplifications were performed
using the LightCycler 2.0 (Roche, Basel, Switzerland) with the
following amplification conditions: 95°C for 15 sec, 58°C for 1
min, 72°C for 1 min and 2 sec for plate reading for 35 cycles.
β-actin was used as the control for normalizing the gene
expression. Each measurement was performed in triplicate. Data
obtained were calculated according to the 2−ΔΔCt method
(14).
 | Table IPrimers for the quantitative
RT-PCR. |
Table I
Primers for the quantitative
RT-PCR.
| Name | Sequence |
|---|
| miR-494 | |
| Sense |
5′-CATAGCCCGTGAAACATACACG-3′ |
| Antisense |
5′-GTGCAGGGTCCGAGGT-3′ |
| CXCR4 | |
| Sense |
5′-GGTGGTCTATGTTGGCGTCT-3′ |
| Antisense |
5′-TGGAGTGTGACAGCTTGGAG-3′ |
| CTNNB1 | |
| Sense |
5′-GCCAGAGCCAACGTCAAGCATCTC-3′ |
| Antisense |
5′-GGCAAAGTGTCCAAAACAAAGCCC-3′ |
| LEF1 | |
| Sense |
5′-TGCTTGCCTGATGACTACCTG-3′ |
| Antisense |
5′-TGAGCACATTTCGGCAATAG-3′ |
| CD44 | |
| Sense |
5′-CTCGATGAGGAAGGGTTTGA-3′ |
| Antisense |
5′-GCAGGAGGAAGAAGAAGCAC-3′ |
| CCND1 | |
| Sense |
5′-ATGTCCCTCGCAAATTGAAG-3′ |
| Antisense |
5′-TCAAAATCGAGATCCCCTTG-3′ |
| E-cadherin | |
| Sense |
5′-TGGAATCCAAGCAGAATTGC-3′ |
| Antisense |
5′-TATGTGGCAATGCGTTCTCTATCCA-3′ |
| N-cadherin | |
| Sense |
5′-TGTTGCTGCAGAAAACCAAG-3′ |
| Antisense |
5′-TTTCACAAGTCTCGGCCTCT-3′ |
| Vimentin | |
| Sense |
5′-CGGGATCCGTCCACCAGGTCCGTGTCCTCG-3′ |
| Antisense |
5′-CCCAAGCTTCTCTTCTTGCAAAGATTCCAC-3′ |
| α-SMA | |
| Sense |
5′-CCGAGATCTCACCGACTACC-3′ |
| Antisense |
5′-TCCAGAGCGACATAGCACAG-3′ |
| β-actin | |
| Sense |
5′-ATTGCCGACAGGATGCAGAA-3′ |
| Antisense |
5′-CAAGATCATTGCTCCTCCTGAGCGCA-3′ |
For miR-494 detection, the miR-494-3p was firstly
obtained by reverse transcription PCR based on the total RNA using
a ReverTra Ace kit (Toyobo Life Science, Shanghai, China). The
primer used for reverse transcription PCR was:
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGAGGTT-3′. The
reverse transcription condition was 42°C for 30 min and 95°C for 5
min. The qRT-PCR primers for miR-494 are listed in Table I. The amplification conditions were
95°C for 10 sec, 55°C for 20 sec, 72°C for 20 sec and 2 sec for
plate reading for 35 cycles.
Western blot analysis
Total proteins were extracted and separated by 12%
SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The bands were
then electroblotted onto a nitrocellulose membrane (Amersham,
Little Chalfont, UK), followed by blocking non-specific binding
using 2% non-fat dry milk in Tris-buffered saline at room
temperature for 2 h. Western blotting was carried out by incubating
the membrane with the following antibodies: anti-CXCR4 (1:1,000;
Abcam, Cambridge, UK), anti-β-actin (1:6,000; KangCheng, Beijing,
China), anti-E-cadherin (1:800), anti-N-cadherin (1:800),
anti-vimentin (1:2,000), anti-α-SMA (1:800) (all from Proteintech
Group), anti-β-catenin (1:1,000), anti-LEF1 (1:800), anti-CD44
(1:800) and anti-cyclin-D1 (1:1,000) (all from Cell Signaling
Technology, Danvers, MA, USA) antibodies diluted in the blocking
buffer overnight at 4°C. The membrane was then incubated with
horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa
Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h at room
temperature. 4-Chloro-1-naphthol (4-CN) was used for protein
visualization.
Dual-luciferase reporter gene assay
The segments of wild-type 3′-UTR of CXCR4 and
mutated 3′-UTR of CXCR4 were obtained according to a previous study
(15). The wild-type 3′-UTR of
CXCR4 or mutated 3′-UTR of CXCR4 was cloned into the downstream of
the luciferase reporter gene in the pGL3 plasmid (Promega, Madison,
WI, USA) by XbaI and NotI restriction sites.
Wild-type 3′-UTR of CXCR4 or mutated 3′-UTR of CXCR4 cloned in the
pGL3 plasmid were co-transfected with the mimic negative control or
miR-494 mimics into HEK293T cells (ATCC) using Lipofectamine 2000.
After a 48-h incubation, the luciferase activity was measured using
the Dual-Luciferase Reporter Assay kit (Promega).
MTT assay
MTT assay was performed to assess cell
proliferation. MDA-MB-231 cells were transfected with miR-494
mimics or inhibitors in 96-well plates (1×104
cells/well). After 12, 24 and 48 h of transfection, culture medium
was replaced with fresh medium containing MTT (Sangon, Shanghai,
China) and incubated with the cells for another 4 h. Then formazan
was dissolved in dimethylsulfoxide (DMSO) (150 μl/well;
Sigma, St. Louis, MO, USA) for 10 min, and the absorbance value at
550 nm in each well was measured with a microplate reader (Thermo
Fisher Scientific, Waltham, MA, USA).
Matrigel invasion assay
A 24-well permeable support plate (Millipore) were
pre-coated with 15 μl of Matrigel (BD Biosciences, Bedford,
MA, USA). Cells (2×104) in 200 μl growth media
were placed onto the top chamber. Normal culture medium containing
100 ng/ml SDF-1 was added to the bottom chamber. Plates were
incubated for 36 h at 37°C. The top of the Matrigel coated
permeable support was rubbed using a cotton swab moistened with
medium to remove the non-invading cells. The cells on the lower
surface of the membrane were fixed with methanol and stained with
crystal violet solution. Invaded cells were counted in 10 random
fields under a microscope.
Statistical analysis
Unless otherwise stated, all experiments were
performed with triplicate samples and repeated at least three
times. Results are presented as mean ± SD. Data are analyzed by the
unpaired Student’s t-test. Statistical comparisons between groups
were performed using one-way ANOVA followed by the Student’s
t-test. Analyses were conducted using SPSS 13.0 software. A value
of P<0.05 was considered to indicate a statistically significant
result.
Results
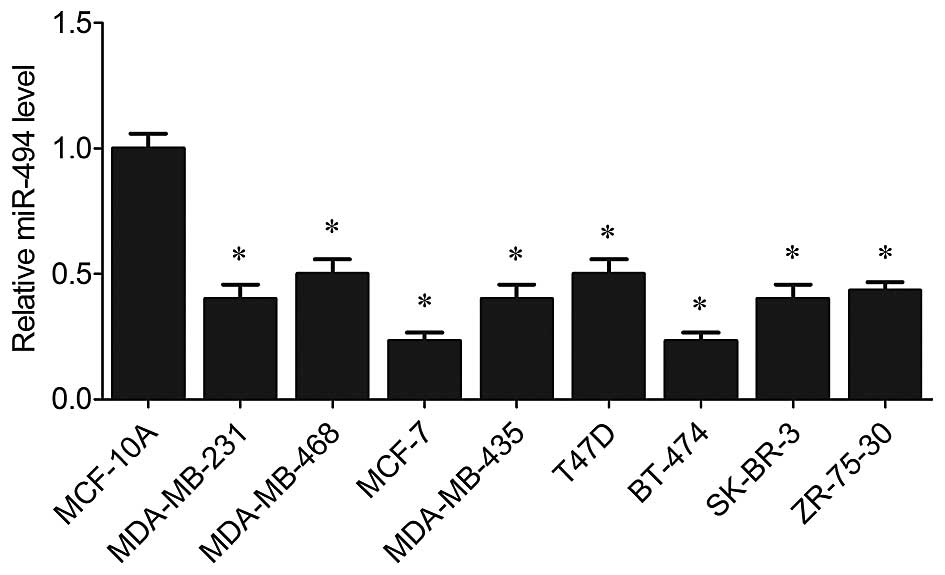
miR-494 is downregulated in human breast
cancer cell lines
Previous studies demonstrated that miR-494 plays
different roles in various types of tumors, either as an oncogenic
miRNA in non-small cell lung cancer and liver tumorigenesis
(10,11), or as an anti-oncogene in human
cholangiocarcinomas, prostate cancer and gastric carcinoma
(12,15,16).
Therefore, expression of miR-494 was assessed in breast cancer cell
lines in the present study. It was revealed by RT-PCR that miR-494
was significantly suppressed in all breast cancer cell lines
(MDA-MB-231, MCF-7, MDA-MB-468, MDA-MB-435, T47D, BT-474, SK-BR-3
and ZR-75-30) compared with the level in the non-malignant breast
epithelial cell line MCF-10 (P<0.05, Fig. 1).
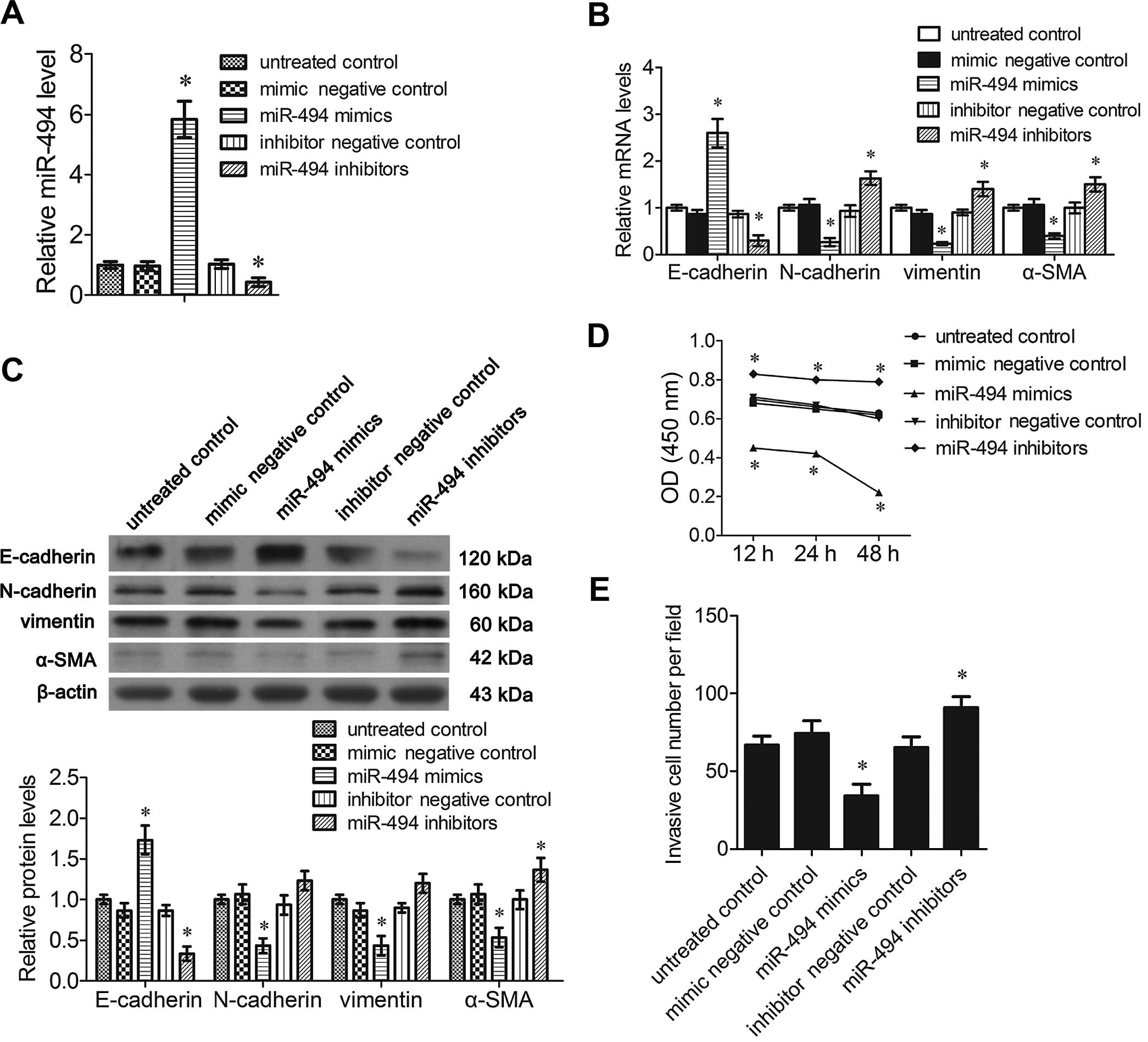
miR-494 mimics suppress EMT,
proliferation and invasion of breast cancer cells
Previous studies have demonstrated that miR-494 is a
potential regulatory miRNA in the EMT of cancer, which is the main
cause of metastasis of breast cancer. The EMT phenotype is dictated
by E-cadherin, N-cadherin, vimentin and α-SMA. To investigate the
association between the expression of miR-494 and EMT of breast
cancer, expression of the EMT markers was detected in the
MDA-MB-231 cells transfected with miR-494 mimics or inhibitors. The
mRNA level of miR-494 was increased significantly following
transfection of the miR-494 mimics in the MDA-MB-231 cells. The
reverse effect was shown in cells transfected with the miR-494
inhibitors (P<0.05, Fig. 2A).
RT-PCR analysis demonstrated that miR-494 mimic transfection
significantly increased the mRNA levels of E-cadherin and decreased
that of N-cadherin, vimentin and α-SMA compared to the untreated
control cells (P<0.05, Fig. 2B).
As expected, in cells transfected with miR-494 inhibitors, the
expression of E-cadherin was significantly decreased, and the
expression of N-cadherin, vimentin and α-SMA was significantly
increased compared to the untreated control cells (P<0.05,
Fig. 2B). Western blot analysis
showed that the expression of these proteins exhibited a similar
trend after transfection with the miR-494 mimics or inhibitors,
except that N-cadherin and vimentin were not significantly affected
by miR-494 inhibitor transfection (P<0.05, Fig. 2C).
MTT assays showed that cells transfected with the
miR-494 mimics showed a significant decrease in cell proliferation
compared to the untreated controls. Cells transfected with the
miR-494 inhibitors showed a small decrease in cell proliferation
(Fig. 2D). The Matrigel invasion
assay was performed to evaluate the invasive ability of the
MDA-MB-231 cells transfected with the miR-494 mimics or inhibitors.
The relative invasive cell number was significantly decreased
following transfection of the miR-494 mimics, and was markedly
increased by miR-494 inhibitor transfection (P<0.05, Fig. 2E).
miR-494 mimics downregulate CXCR4 in
breast cancer cells
The mechanism underlying the inhibitory effect of
miR-494 on the progression of breast cancer is not fully
understood. Previous studies indicated that miR-494 targets CXCR4
to suppress the proliferation, invasion and migration of prostate
cancer (15). Therefore, the
present study determined the effects of miR-494 on the expression
of CXCR4 in the MDA-MB-231 cells. The protein level of CXCR4 was
significantly decreased in the cells transfected with the miR494
mimics compared to the untreated control cells (P<0.05, Fig. 3A), yet the mRNA level of CXCR4 was
not significantly altered (P>0.05, Fig. 3B).
To confirm that miR-494 directly interacts with
CXCR4 mRNA, a dual-luciferase reporter assay was performed.
Wild-type 3′-UTR of CXCR4 or mutated 3′-UTR of CXCR4 (Fig. 3C) constructed in the pGL3 plasmid
were co-transfected with miR-494 mimics into HEK293T cells. Cells
only transfected with wild-type or mutated 3′-UTR of CXCR4 were
used as a control. The results showed that miR-494 mimics
significantly inhibited luciferase activity in the wild-type 3′-UTR
of the CXCR4 co-transfected cells as compared with the control
group (P<0.05, Fig. 3D). Yet,
the reporter constructs with mutated CXCR4 3′-UTR were not affected
by miR-494 mimics (P<0.05, Fig.
3D). This revealed that miR-494 binds to CXCR4 mRNA and
regulated its translation.
miR-494 suppresses the Wnt/β-catenin
pathway via targeting CXCR4
Aberrant activation of the canonical Wnt/β-catenin
pathway plays a critical role in the development of breast cancer.
Previous studies have demonstrated that SDF-1/CXCR4 promotes EMT
and progression of cancer by activation of the Wnt/β-catenin
signaling pathway (4). CXCR4 siRNA
transfection significantly decreased the mRNA and protein levels of
CXCR4 (P<0.05, Fig. 4A and B).
In the MDA-MB-231 cells transfected with CXCR4 siRNA, the mRNA and
protein levels of β-catenin, LEF1, CD44 and cyclin-D1 were
significantly decreased compared to the untreated control
(P<0.05, Fig. 4C and D).
Notably, miR-494 mimics also exhibited the same effect on the
Wnt/β-catenin pathway (P<0.05, Fig.
4C and D). These data suggest that miR-494 suppresses the
Wnt/β-catenin pathway via targeting CXCR4.
Discussion
As the most widespread epithelial tumor among women,
breast cancer has been extensively studied in the past decades.
Previous studies have focused on the EMT of breast cancer cells,
which is the critical factor influencing the invasion and
metastasis of tumors, and leads to a poor prognosis. Previous
studies have demonstrated that miR-494 is a potential regulatory
microRNA in the EMT of cancer. In the present study, significant
upregulation of E-cadherin and downregulation of N-cadherin,
vimentin and α-SMA were observed in the miR-494 mimic-transfected
cells compared to the control cells, indicating suppression of EMT
following miR-494 transfection in breast cancer cells. Notably, the
expression levels of E-cadherin, N-cadherin, vimentin and α-SMA
were not all significantly altered by miR-494 inhibitor
transfection. This may be due to the low baseline of miR-494 in
breast cancer cells.
CXCR4 is a seven-transmembrane G-protein-coupled
receptor for SDF-1. It was reported that SDF-1 signaling through
CXCR4 promotes primary tumor growth and metastasis in breast cancer
and other malignancies (17,18).
It was further found that SDF-1 signaling through CXCR4 promotes
metastasis in breast cancer by increasing angiogenesis and by
establishing an immunosuppressive tumor microenvironment (18). Research in prostate cancer showed
that the constitutive overexpression of miR-494 downregulated the
protein level of CXCR4, leading to suppression of proliferation,
invasion and migration of prostate cancer (15). However, the effect of miR-494 on the
SDF-1/CXCR4 signaling pathway in breast cancer has not yet been
studied. In the present study, we found that miR-494 was markedly
downregulated in breast cancer cell lines compared to the
non-malignant breast epithelial cell line MCF-10A. Furthermore, our
data showed that artificial overexpression of miR-494 suppressed
the expression of CXCR4 post-transcriptionally, which is consistent
with the previous findings in prostate cancer (15). We confirmed that CXCR4 is a target
of miR-494. Furthermore, this effect of miR-494 was found to
inhibit the invasion, EMT and metastasis of breast cancer cells,
which leads to a positive prognosis.
Wingless-related proteins are important regulators
of cell proliferation, differentiation and adhesion (19). The Wnt/β-catenin signaling pathway
plays an important role in the development and regeneration of
several tissues by stimulating the growth of stem cells and
multipotential progenitors (20,21),
particularly in the tumorigenesis of breast cancer by regulating
expression of genes essential for mammary stem cell development.
Wnt ligands, including WNT3A, WNT4, WNT6, WNT8B, WNT9A and WNT10B
were found to be highly expressed in most breast cancer cell lines
(22). These ligands play roles
through the Wnt/β-catenin pathway. The elevated β-catenin levels in
breast cells are supposed to be associated with a predisposition to
breast cancer (22,23). In the present study, we found that
the effect of miR-494 on SDF-1/CXCR4 suppressed the Wnt/β-catenin
pathway, which may be a potential therapeutic target to prevent
breast cancer metastasis and progression.
In conclusion, our findings demonstrated that
miR-494 is downregulated in breast cancer cells, and is able to
suppress cellular proliferation, migration and invasion through
regulation of SDF-1/CXCR4. These results indicate that miR-494 may
serve as a potential therapeutic target for breast cancer.
Acknowledgments
The present study was supported by the Science and
Technology Plan Projects of Social Development Plans for Public
Relations of Shaanxi Province (no. 2010k01-140).
References
|
1
|
Maruani DM, Spiegel TN, Harris EN,
Shachter AS, Unger HA, Herrero-González S and Holz MK: Estrogenic
regulation of S6K1 expression creates a positive regulatory loop in
control of breast cancer cell proliferation. Oncogene.
31:5073–5080. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Malouf GG, Taube JH, Lu Y, Roysarkar T,
Panjarian S, Estecio MR, Jelinek J, Yamazaki J, Raynal NJ, Long H,
et al: Architecture of epigenetic reprogramming following
Twist1-mediated epithelial-mesenchymal transition. Genome Biol.
14:R1442013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gomes LR, Terra LF, Sogayar MC and
Labriola L: Epithelial-mesenchymal transition: Implications in
cancer progression and metastasis. Curr Pharm Biotechnol.
12:1881–1890. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu TH, Yao Y, Yu S, Han LL, Wang WJ, Guo
H, Tian T, Ruan ZP, Kang XM, Wang J, et al: SDF-1/CXCR4 promotes
epithelial-mesenchymal transition and progression of colorectal
cancer by activation of the Wnt/β-catenin signaling pathway. Cancer
Lett. 354:417–426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saxena M, Stephens MA, Pathak H and
Rangarajan A: Transcription factors that mediate
epithelial-mesenchymal transition lead to multidrug resistance by
upregulating ABC transporters. Cell Death Dis. 2:e1792011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Haga CL and Phinney DG: MicroRNAs in the
imprinted DLK1-DIO3 region repress the epithelial-to-mesenchymal
transition by targeting the TWIST1 protein signaling network. J
Biol Chem. 287:42695–42707. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hwang HW, Wentzel EA and Mendell JT: A
hexanucleotide element directs microRNA nuclear import. Science.
315:97–100. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li XH, Qu JQ, Yi H, Zhang PF, Yi HM, Wan
XX, He QY, Ye X, Yuan L, Zhu JF, et al: Integrated analysis of
differential miRNA and mRNA expression profiles in human
radioresistant and radiosensitive nasopharyngeal carcinoma cells.
PLoS One. 9:e877672014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang C, Li C, Li J, Han J, Shang D, Zhang
Y, Zhang W, Yao Q, Han L, Xu Y, et al: Identification of
miRNA-mediated core gene module for glioma patient prediction by
integrating high-throughput miRNA, mRNA expression and pathway
structure. PLoS One. 9:e969082014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Romano F, Garancini M and Uggeri F,
Degrate L, Nespoli L, Gianotti L, Nespoli A and Uggeri F: Surgical
treatment of liver metastases of gastric cancer: State of the art.
World J Surg Oncol. 10:1572012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lim L, Balakrishnan A, Huskey N, Jones KD,
Jodari M, Ng R, Song G, Riordan J, Anderton B, Cheung ST, et al:
MicroRNA-494 within an oncogenic microRNA megacluster regulates
G1/S transition in liver tumorigenesis through
suppression of mutated in colorectal cancer. Hepatology.
59:202–215. 2014. View Article : Google Scholar :
|
|
12
|
Olaru AV, Ghiaur G, Yamanaka S, Luvsanjav
D, An F, Popescu I, Alexandrescu S, Allen S, Pawlik TM, Torbenson
M, et al: MicroRNA down-regulated in human cholangiocarcinoma
control cell cycle through multiple targets involved in the G1/S
checkpoint. Hepatology. 54:2089–2098. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liang Z, Yoon Y, Votaw J, Goodman MM,
Williams L and Shim H: Silencing of CXCR4 blocks breast cancer
metastasis. Cancer Res. 65:967–971. 2005.PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
15
|
Shen PF, Chen XQ, Liao YC, Chen N, Zhou Q,
Wei Q, Li X, Wang J and Zeng H: MicroRNA-494-3p targets CXCR4 to
suppress the proliferation, invasion, and migration of prostate
cancer. Prostate. 74:756–767. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peng C, Zhou K, An S and Yang J: The
effect of CCL19/CCR7 on the proliferation and migration of cell in
prostate cancer. Tumour Biol. 36:329–335. 2015. View Article : Google Scholar
|
|
17
|
Smith MC, Luker KE, Garbow JR, Prior JL,
Jackson E, Piwnica-Worms D and Luker GD: CXCR4 regulates growth of
both primary and metastatic breast cancer. Cancer Res.
64:8604–8612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Orimo A, Gupta PB, Sgroi DC,
Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL
and Weinberg RA: Stromal fibroblasts present in invasive human
breast carcinomas promote tumor growth and angiogenesis through
elevated SDF-1/CXCL12 secretion. Cell. 121:335–348. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wielenga VJ, Smits R, Korinek V, Smit L,
Kielman M, Fodde R, Clevers H and Pals ST: Expression of CD44 in
Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J
Pathol. 154:515–523. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nusse R, Fuerer C, Ching W, Harnish K,
Logan C, Zeng A, ten Berge D and Kalani Y: Wnt signaling and stem
cell control. Cold Spring Harb Symp Quant Biol. 73:59–66. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Benhaj K, Akcali KC and Ozturk M:
Redundant expression of canonical Wnt ligands in human breast
cancer cell lines. Oncol Rep. 15:701–707. 2006.PubMed/NCBI
|
|
23
|
Schlange T, Matsuda Y, Lienhard S, Huber A
and Hynes NE: Autocrine WNT signaling contributes to breast cancer
cell proliferation via the canonical WNT pathway and EGFR
trans-activation. Breast Cancer Res. 9:R632007. View Article : Google Scholar
|