Introduction
Soft tissue sarcomas are tumors arising from
mesenchymal cell precursors that are committed towards the
morphogenesis of soft tissues such as fat, muscle and deep skin
tissues. Rhabdomyosarcoma (RMS) is considered a myogenic tumor and
is classified as the most frequent sarcoma affecting children and
adolescents (1). The current
classification defines five different histotypes, with embryonal
(eRMS) and alveolar (aRMS) subsets being the most frequently
observed in children <5 years and in adolescents, respectively
(2,3). The eRMS variant is the most treatable
and most common subtype representing ~80% of RMS, while aRMS is
more aggressive and characterized by a poorer prognosis. The
genetic alterations characterizing eRMS commonly involve the loss
of heterozygosis on chromosome region 11p15.5 (4), gain of chromosomes (5,6) and
mutations on genes involved with growth factor signaling pathways
(7–15). This leads to uncontrolled cell
growth and the interruption of proper myogenic differentiation.
Conversely, the aRMS subset is commonly characterized by the
expression of Pax3-forkhead box O1 (FOXO1), a fused transcription
factor derived from the chromosomal translocation t(2;13)(q35;q14),
which juxtaposes the DNA-binding domain of Pax3 to the potent
transactivation domain of FOXO1 (16). In the absence of the original Pax3
transactivation domain, the chimeric protein drives in a
constitutive manner the transcription of numerous genes involved in
muscle embryogenesis, such as c-MET and FGFR4,
essentially maintaining the muscle precursors in a long-lasting
proliferative state and thereby facilitating tumor initiation,
aggressiveness and metastatic ability (16–19).
Melatonin is a small molecule derived from
tryptophan metabolism and secreted by the pineal gland during
periods of darkness (20,21). Melatonin is involved in the
regulation of seasonal and circadian rhythms, effects that are
mediated by melatonin binding to specific receptors, such as MT1
and MT2, which in turn trigger the downstream activation of CLOCK
and BMAL1 factors driving a complex transcriptional program
(22,23). Besides regulating the rhythm
adaptations, melatonin exhibits additional functions, including the
promotion of cell survival, neuroprotection and cardioprotection
likely due to antioxidant properties (24–27).
Notably, previous findings have shown a role for melatonin in
preventing tumor initiation and progression (28–34).
Specifically, melatonin was reported to inhibit cell proliferation
and induce apoptosis in osteosarcoma (35), B-lymphoma (36) and colorectal cancer cells (37), as well as to decrease the weight of
tumor masses in breast and prostate cancer (38–40).
Based on those reports, the present study was conducted to examine
the effects of melatonin on RMS cell lines in vitro. For
this purpose, we employed human RMS cell lines as well as primary
mouse tumor cultures established from transgenic mice (41) to evaluate the effects of melatonin
on cell viability, proliferation and differentiation. In
vitro assays and morphological analysis using electronic and
confocal microscopy were performed.
Materials and methods
Reagents
Reagents were purchased from Sigma-Aldrich (Milan,
Italy), unless otherwise stated. Cell culture materials were
purchased from Jet-Biofil (Carlo Erba Reagents-Dasit Group,
Cornaredo, Italy).
Human cell lines and primary mouse tumor
cultures
Human RD (eRMS) and RH30 (aRMS) cells were purchased
from the European Collection of Cell Cultures (ECACC; Salisbury,
UK). The primary tumor mouse cultures, U57810 (eRMS) and U23674
(aRMS), were established from transgenic mice (41). In particular, eRMS mouse models were
generated by crossing p53- or Ptch1-deficient conditional mice with
Myf6-Cre mice to achieve the deletion of p53 with or without
concurrent Ptch1 deletion in differentiating Myf6-positive
myoblasts. The Myf6Cre/p53−/− mouse strain was
characterized by the highest percentage of eRMS (41). aRMS mouse models were characterized
by knockout alleles of p53 or INK4a/ARF locus with concomitant
Pax3-FOXO1 knock-in allele, which were restricted to
differentiating Myf6-positive myoblasts, resulting in the
Myf6Cre/Pax3-FOXO1/p53−/− mouse strain (41).
Cell culture conditions
Cells were maintained at 37°C and 5% CO2
in a humidified incubator and cultured in a growth medium (GM)
comprising high-glucose Dulbecco’s modified Eagle’s medium (DMEM)
(D6429) supplemented with 10% fetal bovine serum (FBS) (FA30A15101;
Carlo Erba, Milan, Italy) in the presence of 100 µg/ml
penicillin/streptomycin (A5955) and 1% L-glutamine (G7513) (only
for RH30 cells). To induce myodifferentiation, 80% confluent cells
were switched to differentiation medium (DM) comprising DMEM
supplemented with 2% horse serum (H1270).
Pharmacological treatments
Cells were treated with different concentrations
(0.01, 0.1, 1, and 2 mM) of melatonin (461326) or vehicle alone
[dimethylsulfoxide (DMSO)] (D5879). The cells were also treated
with melatonin in the absence or presence of chemotherapeutic drugs
such as doxorubicin (0.15 ng/ml) (D1515) and cisplatin (2
µg/ml) (P4394), which were previously diluted in
H2O and DMSO, respectively.
Cell proliferation assay
eRMS and aRMS cells were seeded in 24-well plates at
a density of 10×103 and 15×103, respectively.
After 24, 48 and 72 h of melatonin treatment, the cells were
harvested, fixed in paraformaldehyde (F8775) and stained for 10 min
with crystal violet (C0075) solution [0.2% in phosphate-buffered
saline (PBS) (D8537) with 20% methanol (32213)]. The samples were
then collected in 600 µl of SDS (74255) solution (1% in PBS)
and absorbance of the total homogenates, as measured by reading the
plate at 540 nm emission wavelengths, was proportional to the
amount of viable and proliferating cells that incorporated the
crystal violet. In addition, the cell proliferation was expressed
as the growth rate, which was calculated using Microsoft Excel 2010
software. The results were representative of at least three
independent experiments.
Cell viability assay
A neutral red assay was employed to determine the
percentage of viable cells that incorporated the neutral red dye in
lysosomes, as initially described by Borenfreund and Puerner
(42), a protocol subsequently
modified by Repetto et al (43). Briefly, the cells were seeded in
96-well plates at a density of 1.5×103. After 24, 48 and
72 h of melatonin treatment, the cells were incubated for 2 h with
neutral red dye (40 µg/ml) (N7005) dissolved in DMEM with 5%
FBS. After washing the cells with PBS, 150 µl of neutral red
destaining solution [50% ethanol (02860), 49% deionized water, and
1% glacial acetic acid (100015N; BDH Laboratory Supplies,
Dawsonville, GA, USA)] was added, followed by gentle agitation for
10 min, until complete dissolution was achieved. Absorbance was
then measured by reading the plate at 540 nm emission wavelengths.
The results were analyzed using Microsoft Excel 2010 software and
presented as the percentage of control values. Images of cell
viability assays showed representative results of at least three
independent experiments.
Immunoblotting analysis
Protein homogenates were obtained by harvesting
cells in a cold RIPA lysis buffer, comprising 20 mM Tris-HCl (ph
7.6) (T1503), 1% Nonidet P40 (NP40), 0.5% sodium deoxycholate
(D6750), 0.1% SDS (74255), 50 mM NaCl (S7653) and a cocktail of
protease inhibitors (1836153; Roche, Milan, Italy) plus phosphatase
inhibitors [1 mM Na3VO4 (S6508) and 4 mM NaF
(S7920)]. The protein concentration was calculated by a Bradford
reagent (B6916) assay and an equal amount of protein samples was
separated by SDS-PAGE under reducing conditions and transferred to
polyvinylidene fluoride membranes (P2938). Incubation with specific
primary antibodies was followed by peroxidase-conjugated secondary
antibodies (goat polyclonal anti-mouse IgG-HRP sc-2005; from Santa
Cruz Biotechnology, Inc., Dallas, TX, USA; donkey polyclonal
anti-rabbit IgG no. 31458; from Thermo Scientific, Erembodegem,
Belgium) and the resulting immune complexes were visualized using
the enhanced chemiluminescence reagent (STS-E 500; GeneSpin, Milan,
Italy). Immune-reactive bands were quantified using densitometry
analyses (Software Gel-Pro Analyzer, version 4).
Antibodies
The antibodies were purchased from Santa Cruz
Biotechnology, Inc., unless otherwise stated. The primary
antibodies used were: mouse monoclonal anti-myosin heavy chain,
(sc-32732; 1:1,000 dilution); mouse monoclonal anti-caveolin-3
(610420; 1:1,000 dilution; BD, Buccinasco, Italy); rabbit
polyclonal anti-caspase-3 (H-277) (sc-7148; 1:500 dilution; Cell
Signaling, Milan, Italy); rabbit polyclonal anti-Bax (sc-526; 1:500
dilution); rabbit polyclonal anti-Bcl-2 (sc-492; 1:500 dilution);
and mouse monoclonal anti-α-tubulin (T5168; 1:10,000 dilution;
Sigma-Aldrich).
Scanning electron microscopy (SEM)
RH30 cells were cultured and treated directly on
coverslips in Petri dishes. After washing with 0.1 M phosphate
buffer, adherent and suspended cells were fixed with 2.5%
glutaraldehyde (G5882) in 0.1 M phosphate buffer for 1 h. The
suspended cells adhered to polylysine-coated coverslips. The
samples were post-fixed with 1% osmium tetroxide (OsO4)
(O021; Strumenti, Roma, Italy) in 0.1 M phosphate buffer for 1 h.
After alcohol dehydration, the samples were critical point dried,
gold sputtered and observed using a Philips 515 scanning electron
microscope (FEI, Italy) (44).
Transmission electron microscopy
(TEM)
RH30-treated cells were washed and fixed with 2.5%
glutaraldehyde (G5882) in 0.1 M phosphate buffer for 15 min. The
cells were scraped and centrifuged at 300 × g for 10 min. The
pellets were fixed in 2.5% glutaraldehyde for an additional 30 min.
The suspended cells were collected in Eppendorf, centrifuged and
fixed for 45 min in glutaraldehyde. The samples were post-fixed in
1% OsO4 (O021) for 1 h, alcohol dehydrated and embedded
in araldite (02860) (45). Thin
sections were stained with uranyl acetate and lead citrate and
analyzed using a Philips CM10 transmission electron microscope
(FEI).
Confocal microscopy fluorescence
Adherent cells were cultured and treated directly on
coverslips in Petri dishes. The suspended cells were collected in
Eppendorf, fixed in 4% paraformaldehyde (F8775) for 30 min and then
plated on polylysine-coated coverslips. The cells were then fixed
in 4% paraformaldehyde in PBS (ph 7.4) for 30 min and washed twice
using PBS. The cells were then pre-treated with RNase A (10
µg/ml) (12091-021) in PBS for 30 min and exposed to an equal
mixture of propidium iodide (PI; 1 µg/ml) (P3566) and
acridine orange (AO; 1 µg/ml) (A3568) (all from Life
Technologies, Monza, Italy) in PBS at room temperature in the dark
for 10 min.
AO and PI are intercalating fluorochromes that emit
green and red fluorescence, respectively, when they are bound to
DNA. Only AO diffuses through the plasma membrane of both viable
and early apoptotic cells. Viable cells exhibit a green nucleus
with intact structure, while apoptotic cells exhibit a bright-green
nucleus, with condensation of chromatin. PI only enters late
apoptotic and necrotic cells, resulting in double staining with AO
and PI (45). The samples were
observed with a Leica TCS-SP5 CLSM connected to a DMI 6000 CS
inverted microscope (Leica Microsystems CMS Gmbh; AO and PI
excitation were at 488 and 500 nm, respectively, and their emission
signals were detected at 617 and 525 nm, respectively).
Statistical analysis
The differences between the groups were analyzed by
the unpaired Student’s t-test and one-way ANOVA test (with Dunnet’s
post-hoc test), using Prism 4 software for Windows (GraphPad
Software, San Diego, CA, USA). P<0.05 was considered to indicate
a significant result.
Results
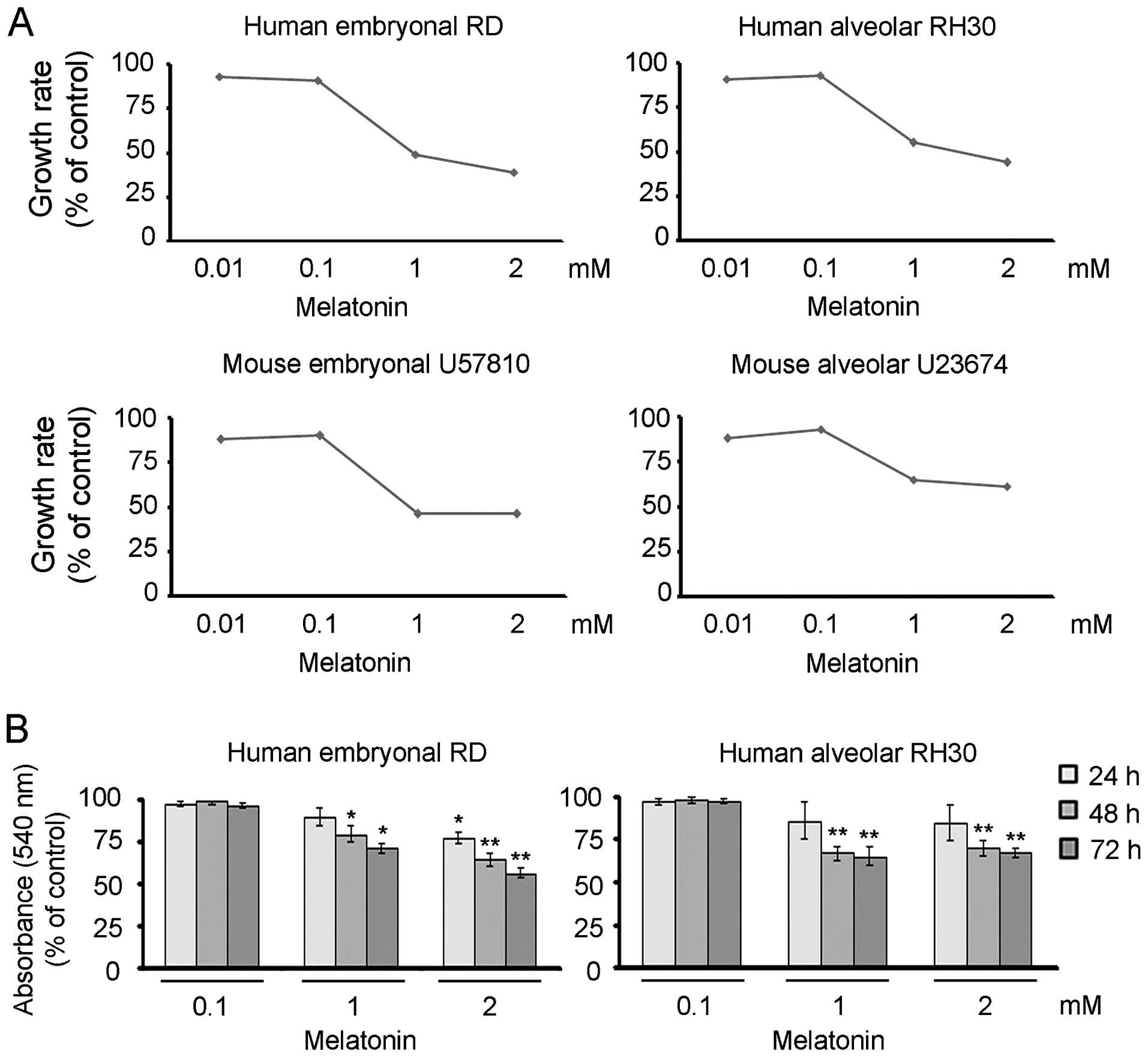
Melatonin suppresses cell proliferation
and triggers apoptotic and necrotic features in RMS cells
We evaluated whether melatonin administration would
influence the cell growth of the human RMS cell lines (i.e.
embryonal RD and alveolar RH30) and primary mouse tumor cultures
(i.e., embryonal U57810 and alveolar U23674). For this purpose, the
proliferation of cells that received melatonin once was determined
over a time-course of 72 h by means of crystal violet assay.
Treating different lines with increasing concentrations of
melatonin, ranging from 0.01 to 2 mM, led to a significant
impairment of cell proliferation starting from a dose of 1 mM in
comparison to vehicle-treated cells, as indicated after calculation
of the growth rate (Fig. 1A). To
determine whether the melatonin effects were attributable to the
inhibition of cell proliferation rather than impaired cell
viability, we performed the neutral red assay using the cell lines
under the same experimental conditions. As shown in Fig. 1B, 72 h of exposure with a
concentration of 1 or 2 mM triggered the loss of ~50% of RMS cells
in comparison to vehicle-treated cells, indicating that melatonin
has cytotoxic effects. This latter result was confirmed by
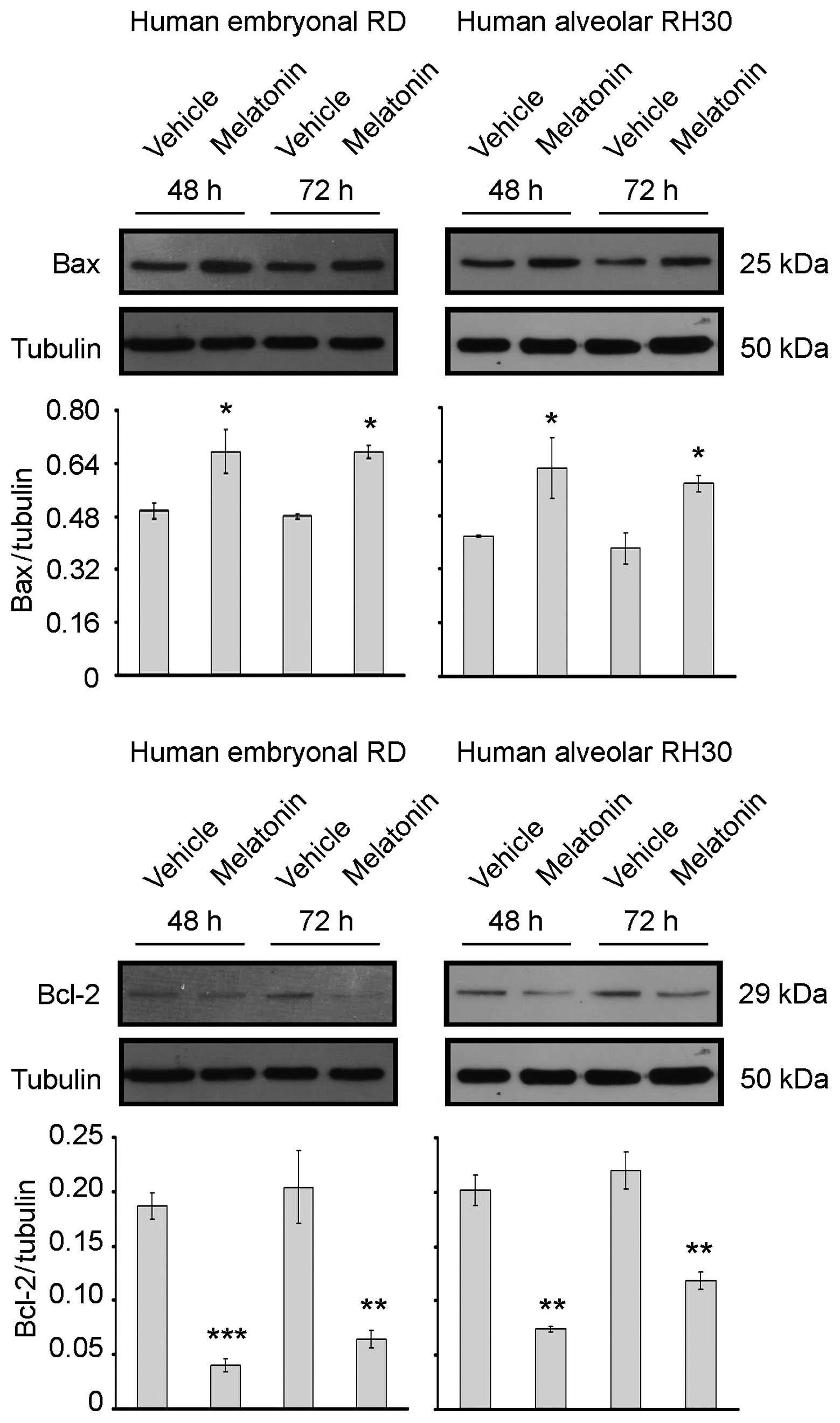
immunoblotting analysis of Bax and Bcl-2 expression, two proteins
that can be either pro- or anti-apoptotic, respectively (46–48).
As shown in Fig. 2,
treatment of RD and RH30 cells with 1 mM melatonin promoted an
increase in the pro-apoptotic Bax expression, while the expression
levels of anti-apoptotic Bcl-2 were downregulated in comparison to
those in untreated cells. These results indicated that melatonin,
not only behaves as a cytostatic factor on RMS cell growth, but
also impairs the survival of different RMS lines by triggering an
apoptotic program. Thus, at the ultrastructural level, the
pro-apoptotic effects of melatonin, RH30 cells treated or untreated
were analyzed by electronic and confocal microscopy.
Control cells showed an obvious healthy morphology
characterized by the presence of intact subcellular structures, as
observed by means of SEM (Fig. 3A)
and TEM (Fig. 3B and C),
respectively. In addition, AO/PI double staining showed a uniform
green labeling suggestive of cellular healthy structures (Fig. 3D). After 1 mM melatonin for 24 h a
heterogeneous situation developed: some cells maintained good cell
viability similar to the control condition, while other cells
showed a round apoptotic-like morphology (Fig. 3E and F). In particular, TEM analyses
revealed some cells with an intense chromatin condensation, a
typical apoptotic pattern (Fig.
3G). At the confocal microscopy level some cells appeared
rounded and early apoptotic features were evident (Fig. 3H). After 48 h, melatonin-treated
cells were almost all detached showing a round apoptotic morphology
while only a small number of adherent cells exhibited an atrophic
behavior, due to cytoplasm shrinkage (Fig. 3I). In addition, rounded cells
suggestive of necrotic features also appeared (Fig. 3J). These cells were characterized by
typical apoptotic features, including the presence of chromatin
condensation, cytoplasm vacuolization and secondary necrosis as
confirmed by TEM analysis (Fig.
3K). Consistent with this, an increased number of apoptotic
cells showed orange areas due to PI permeability suggestive of
cells in late apoptosis (Fig. 3L).
After 72 h, the melatonin-treated cells observed at the SEM level
exhibited apoptotic and necrotic features, being completely
detached and showing a rounded morphology, with disruption of cell
membranes in those that were necrotic (Fig. 3M and N). As shown by TEM, necrotic
cells were characterized by cytoplasmic vacuolization due to
membrane disruption and loss of cell components during the necrotic
process (Fig. 3Q). The few adherent
apoptotic cells exhibited large orange areas (Fig. 3P, inset) as observed during late
apoptosis, whereas suspended cells showed bright-green nuclei
predictive of apoptotic bodies (Fig.
3P). RH30 cells treated with 2 mM melatonin were characterized
by round, blebbed cells after 24 and 48 h (Fig. 3Q and R, respectively). TEM analysis
at 48 h of treatment highlighted dark areas predictive of DNA
condensation and cytoplasmic vacuolization (Fig. 3S). Using AO and PI double-staining,
melatonin-treated cells showed orange staining at the nuclei due to
late apoptosis and necrosis already after 24 h (Fig. 3T), as also observed after 48 h (data
not shown).
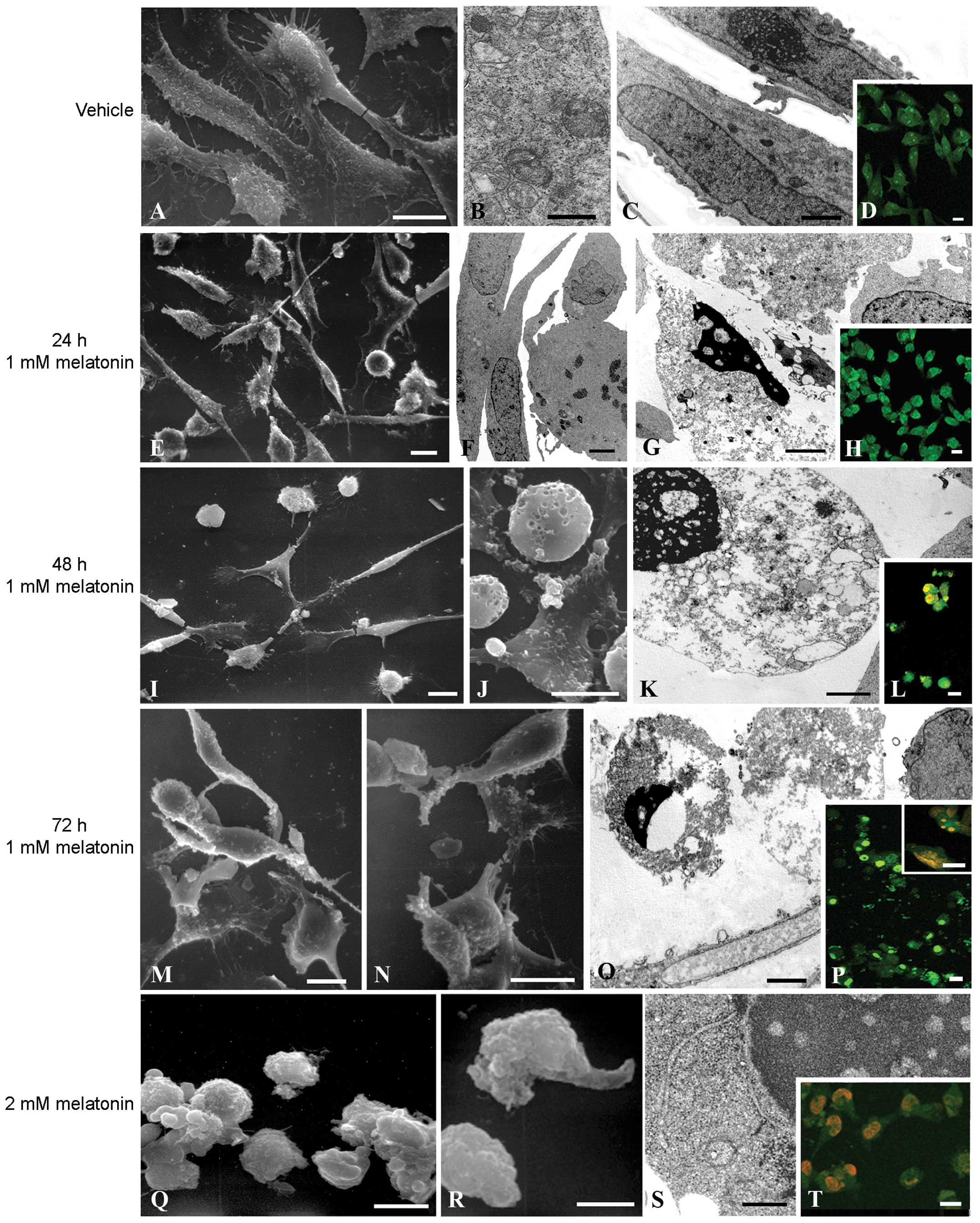 | Figure 3Microscopy analysis of the
morphological changes occurring in melatonin-treated RH30 cells.
Untreated control cells were observed using (A) SEM, (B and C) TEM
and (D) CLSM. (A) Control cells showed adherent morphology. (B)
Mitochondrion with healthy structure. (C) Well-defined intact
nucleus. (E) AO green staining of vital cells. Bars, A and D, 10
micron; C, 2 micron; and B, 500 nm. Cytotoxic effect of melatonin
on RH30 cells. RH30 cells were treated with melatonin at a
concentration of 1 mM for 24, 48 and 72 h and observed at (E, I, J,
M and N) SEM, (F, G, K and O) TEM and (H, L and P) CLSM. (E)
RH30-treated cells with an elongated morphology. (F) Intact nucleus
of a vital cell. (G) Adherent cell showing an apoptotic body. (H)
AO green staining of adherent cells. (I) Adherent cells with an
elongated morphology with some apoptotic rounded cells. (J) Rounded
apoptotic cells. (K) Apoptotic cells showing an apoptotic body and
a well-defined plasma membrane. (L) Orange staining due to PI
permeability in apoptotic cells. (M and N) Necrotic cells with
disrupted cell membranes. (O) Necrotic cells showing disruption of
cell membranes and cytoplasmic vacuolization. (P) AO bright-green
staining in early apoptotic cells and PI orange staining in late
apoptotic and necrotic cells. Bars: E, H, I, J, L, M, N and P, 10
micron; F, G, K and O, 500 nm. Cytotoxic effect of melatonin on
RH30 cells. RH30 cells were treated with 2 mM melatonin for 24 and
48 h and observed at (Q and R) SEM, (S) TEM and (T) CLSM. (Q and R)
Necrotic-treated cells with disrupted cell membranes. (S) Necrotic
cell showing DNA condensation and disruption of cell membranes. (T)
PI orange staining in apoptotic and necrotic cells. Bars: Q, R and
T, 10 micron; S, 500 nm. SEM, scanning electron microscopy; TEM,
transmission electron microscopy; AO, acridine orange; PI,
propidium iodide. |
Melatonin impairs the myogenic
differentiation in embryonal RD cells
Forced differentiation of tumor cells induced by
anticancer agents has been widely exploited to limit the growth of
tumor masses. In this regard, melatonin has been hypothesized to
promote a differentiated phenotype in some tumors, such as gastric
(49) and prostate cancer (50). Thus, to understand whether melatonin
influences the myogenic differentiation of RMS, we employed human
RD cells, which commonly exhibit a consistent myogenic potential in
comparison to alveolar RH30 cells. For RD cells, the DM in the
absence or presence of melatonin was replaced daily using different
concentrations of melatonin. The extent of myogenic differentiation
reached by cells in the different conditions was measured by
immunoblotting analysis of markers that are normally increased
during the differentiation of myoblasts, including caveolin-3
(Cav-3) and myosin heavy chain (MHC). Melatonin treatment led to a
dose-dependent impairment of myogenic differentiation, since Cav-3
and MHC levels were reduced in comparison to the controls. In
particular, melatonin at a concentration of 1 mM completely
abolished the myogenic differentiation in RMS cells, as both Cav-3
and MHC levels were undetectable (Fig.
4A). We also administered melatonin to RD cells after which
they were differentiated for 4 days and then analyzed for caspase-3
proteolytic activation, which is commonly utilized as a readout of
the cell apoptotic program. As observed by immunoblotting analysis,
melatonin-treated differentiated RD cells were characterized by
increased levels of caspase-3 cleaved fragments (~19 and 17 kDa) in
comparison to vehicle-treated cells after treatment for 24 and 48 h
(Fig. 4B). These experiments
demonstrated that melatonin has no positive effects on RMS
differentiation, but behaves as a cytotoxic drug by triggering a
caspase-dependent apoptosis.
Melatonin sensitizes RD and RH30 cells to
cell death induced by chemotherapeutic agents doxorubicin and
cisplatin
Previous findings have suggested that combination
therapies including melatonin and conventional cancer drugs enhance
success by increasing drug efficacy while reducing their side
effects. In most clinical trials where melatonin was used in
conjunction with chemotherapeutic drugs, improved overall survival
and patient conditions were observed (51,52).
This suggests that melatonin enhances the efficacy of chemotherapy
and reduces side effects (53,54). A
combination of melatonin and doxorubicin was reported to enhance
the growth inhibitory effect and induction of apoptosis in human
hepatoma cells in comparison to melatonin or doxorubicin used alone
(55). To verify the combined
effects of melatonin and chemotherapy drugs on RMS chemoresistance,
RD and RH30 cells were concurrently treated with 1 mM melatonin and
0.15 ng/ml doxorubicin or 2 µg/ml cisplatin for up to 48 h.
As shown in Fig. 5, the two
chemotherapies were effective in reducing the proliferation of RMS
cell lines in comparison to the controls, as observed after 48 h of
treatment. In addition, the concomitant treatment of melatonin with
doxorubicin for up to 48 h produced a synergistic inhibitory effect
(Fig. 5A). Similar results were
obtained following treatment with melatonin and cisplatin (Fig. 5B). These results demonstrate a
possible role of melatonin in improving the chemotherapy
efficacy.
Discussion
Results obtained in the present study indicate that
melatonin, when used at concentrations varying from 0.01 to 2 mM,
profoundly affect the cell survival of rhabdomyosarcoma (RMS), the
most frequent myogenic sarcoma affecting children and adolescents
(4). In human cell lines
representative of the most frequent RMS categories, i.e., the eRMS
and aRMS subtypes, we observed that melatonin limited cell
proliferation and triggered morphological and subcellular changes
typically recognizable in apoptotic cells, such as DNA
fragmentation, disruption of cell membranes and proteolytic
cleavage of caspases. We also observed similar effects in primary
mouse tumor RMS cultures which, having been derived from mice with
specific genetic backgrounds, faithfully recapitulate the onset of
RMS genesis (41). In the cell
cultures, the ability of melatonin to increase apoptosis was not
exclusively correlated with cell cycle-dependent effects, since we
observed melatonin to be effective in triggering cell death even in
RMS cells that had withdrawn from the cell cycle to attempt
differentiation. These observations suggest a potential efficacy of
melatonin towards undifferentiated and more differentiated tumor
histotypes. Studies on cancer cells have shown that
antiproliferative and pro-apoptotic effects of melatonin were
achieved with high doses, as compared with those detected in the
blood at night. However, it is known that the intracellular levels
of melatonin may be much higher than in blood (56). To explain the reason for melatonin
often requiring to be added at pharmacological concentrations to
produce inhibitory effects, a regulatory mechanisms by which its
accumulation in cell membranes acts as a reservoir, limiting the
net amount of the biological active indoleamine has been suggested
(57).
Previous findings have indicated that melatonin
produces no consistent adverse effects over various concentrations
(58), suggesting that it may be
useful to improve the efficacy of conventional cytotoxic agents. In
this regard, we showed that melatonin synergized with
chemotherapeutic drugs in human RMS cell lines. To the best of our
knowledge, this is the first study showing indolamine to be
effective in the enhancement of cell death on myogenic tumor cells
using a complementary approach with doxorubicin or cisplatin drugs,
as already observed in clinical trials on different types of cancer
(51–53,59).
The mechanisms underlying the effect of melatonin on
apoptosis have not been clarified and appear to be, to some extent,
context-specific (60–62). Some melatonin actions are mediated
by specific membrane receptors, known as MT1 and MT2, that are
known to be expressed in RMS tumor samples as demonstrated using
tissue microarray analysis (data not shown). However, high doses of
melatonin were found to be effective in inhibiting proliferation.
Thus, RMS cells likely have a non-receptor-mediated action, since
melatonin has been shown to even permeate into cells by means of
receptor-independent processes. Notably, differentiated RD cells
seemingly exhibited a marked responsiveness to melatonin, since
already at 0.01 and 0.1 mM concentrations we observed a negative
effect on myogenic differentiation. In this context, whether the
expression levels of melatonin receptors may differ between
proliferating and differentiated cells, thus accounting for the
different observed sensitivities should be investigated. Whereas in
normal cells melatonin and its metabolites act as efficient radical
scavengers (63), it has been
suggested that changes in the oxidative status account for the
ability of melatonin to induce apoptosis in cancer cells (62,64,65).
In this regard, a correlation between the increase in ROS
production and the induction of melatonin-driven apoptosis has been
reported in several cell lines (66). Consistent with this evidence, we
have preliminarily observed a reduction in the melatonin cytotoxic
effect by pretreating RMS cells with vitamin E (data not shown), a
lipid-soluble antioxidant molecule. Although these observations are
under investigation, it remains to be established whether the
potential changes in the redox status is the cause rather than the
consequence of the increased cell death.
In conclusion, the molecular mechanisms underlying
the cytotoxicity on RMS cells, as observed for other types of
cancer, deserve attention for establishing whether a rationale
occurs for the introduction of melatonin as an adjuvant in the
multimodality approach currently used against RMS.
Acknowledgments
We are grateful to Charles Keller (Oregon Health and
Science University, USA) for providing the primary mouse tumor
cultures of RMS.
Abbreviations:
|
FGFR4
|
fibroblast growth factor receptor
4
|
|
FOXO1
|
forkhead box O1
|
|
Myf6
|
myogenic factor 6
|
|
Pax3 or -7
|
paired box 3 or -7
|
References
|
1
|
Saab R, Spunt SL and Skapek SX: Myogenesis
and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Curr
Top Dev Biol. 94:197–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dasgupta R and Rodeberg DA: Update on
rhabdomyosarcoma. Semin Pediatr Surg. 21:68–78. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Parham DM, Alaggio R and Coffin CM:
Myogenic tumors in children and adolescents. Pediatr Dev Pathol.
15(Suppl 1): S211–S238. 2012. View Article : Google Scholar
|
|
4
|
Ognjanovic S, Linabery AM, Charbonneau B
and Ross JA: Trends in childhood rhabdomyosarcoma incidence and
survival in the United States, 1975–2005. Cancer. 115:4218–4226.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Williamson D, Missiaglia E, de Reyniès A,
Pierron G, Thuille B, Palenzuela G, Thway K, Orbach D, Laé M,
Fréneaux P, et al: Fusion gene-negative alveolar rhabdomyosarcoma
is clinically and molecularly indistinguishable from embryonal
rhabdomyosarcoma. J Clin Oncol. 28:2151–2158. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen X, Stewart E, Shelat AA, Qu C,
Bahrami A, Hatley M, Wu G, Bradley C, McEvoy J, Pappo A, et al: St.
Jude Children’s Research Hospital-Washington University Pediatric
Cancer Genome Project: Targeting oxidative stress in embryonal
rhabdomyosarcoma. Cancer Cell. 24:710–724. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shukla N, Ameur N, Yilmaz I, Nafa K, Lau
CY, Marchetti A, Borsu L, Barr FG and Ladanyi M: Oncogene mutation
profiling of pediatric solid tumors reveals significant subsets of
embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in
growth signaling pathways. Clin Cancer Res. 18:748–757. 2012.
View Article : Google Scholar :
|
|
8
|
Shern JF, Chen L, Chmielecki J, Wei JS,
Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, et
al: Comprehensive genomic analysis of rhabdomyosarcoma reveals a
landscape of alterations affecting a common genetic axis in
fusion-positive and fusion-negative tumors. Cancer Discov.
4:216–231. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abraham J, Prajapati SI, Nishijo K,
Schaffer BS, Taniguchi E, Kilcoyne A, McCleish AT, Nelon LD, Giles
FG, Efstratiadis A, et al: Evasion mechanisms to Igf1r inhibition
in rhabdomyosarcoma. Mol Cancer Ther. 10:697–707. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taylor JG VI, Cheuk AT, Tsang PS, Chung
JY, Song YK, Desai K, Yu Y, Chen QR, Shah K, Youngblood V, et al:
Identification of FGFR4-activating mutations in human
rhabdomyosarcomas that promote metastasis in xenotransplanted
models. J Clin Invest. 119:3395–3407. 2009.PubMed/NCBI
|
|
11
|
Crose LE and Linardic CM: Receptor
tyrosine kinases as therapeutic targets in rhabdomyosarcoma.
Sarcoma. 2011:7569822011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee Y, Kawagoe R, Sasai K, Li Y, Russell
HR, Curran T and McKinnon PJ: Loss of suppressor-of-fused function
promotes tumorigenesis. Oncogene. 26:6442–6447. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Petricoin EF III, Espina V, Araujo RP,
Midura B, Yeung C, Wan X, Eichler GS, Johann DJ Jr, Qualman S,
Tsokos M, et al: Phosphoprotein pathway mapping: Akt/mammalian
target of rapamycin activation is negatively associated with
childhood rhabdomyosarcoma survival. Cancer Res. 67:3431–3440.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guenther MK, Graab U and Fulda S:
Synthetic lethal interaction between PI3K/Akt/mTOR and Ras/MEK/ERK
pathway inhibition in rhabdomyosarcoma. Cancer Lett. 337:200–209.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hahn H, Wojnowski L, Specht K, Kappler R,
Calzada-Wack J, Potter D, Zimmer A, Müller U, Samson E,
Quintanilla-Martinez L, et al: Patched target Igf2 is indispensable
for the formation of medulloblastoma and rhabdomyosarcoma. J Biol
Chem. 275:28341–28344. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marshall AD and Grosveld GC: Alveolar
rhabdomyosarcoma-The molecular drivers of PAX3/7-FOXO1-induced
tumorigenesis. Skelet Muscle. 2:252012. View Article : Google Scholar
|
|
17
|
Barr FG, Galili N, Holick J, Biegel JA,
Rovera G and Emanuel BS: Rearrangement of the PAX3 paired box gene
in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat
Genet. 3:113–117. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Graf Finckenstein F, Shahbazian V,
Davicioni E, Ren YX and Anderson MJ: PAX-FKHR function as pangenes
by simultaneously inducing and inhibiting myogenesis. Oncogene.
27:2004–2014. 2008. View Article : Google Scholar
|
|
19
|
Keller C and Guttridge DC: Mechanisms of
impaired differentiation in rhabdomyosarcoma. FEBS J.
280:4323–4334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carlberg C: Gene regulation by melatonin.
Ann NY Acad Sci. 917:387–396. 2000. View Article : Google Scholar
|
|
21
|
Stehle JH, Saade A, Rawashdeh O, Ackermann
K, Jilg A, Sebestény T and Maronde E: A survey of molecular details
in the human pineal gland in the light of phylogeny, structure,
function and chronobiological diseases. J Pineal Res. 51:17–43.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hardeland R: Melatonin: Signaling
mechanisms of a pleiotropic agent. Biofactors. 35:183–192. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Challet E: Minireview: Entrainment of the
suprachiasmatic clockwork in diurnal and nocturnal mammals.
Endocrinology. 148:5648–5655. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luchetti F, Betti M, Canonico B,
Arcangeletti M, Ferri P, Galli F and Papa S: ERK MAPK activation
mediates the antiapoptotic signaling of melatonin in UVB-stressed
U937 cells. Free Radic Biol Med. 46:339–351. 2009. View Article : Google Scholar
|
|
25
|
Luchetti F, Canonico B, Betti M,
Arcangeletti M, Pilolli F, Piroddi M, Canesi L, Papa S and Galli F:
Melatonin signaling and cell protection function. FASEB J.
24:3603–3624. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tengattini S, Reiter RJ, Tan DX, Terron
MP, Rodella LF and Rezzani R: Cardiovascular diseases: Protective
effects of melatonin. J Pineal Res. 44:16–25. 2008.
|
|
27
|
Zhang HM and Zhang Y: Melatonin: A
well-documented antioxidant with conditional pro-oxidant actions. J
Pineal Res. 57:131–146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bukowska A: Anticarcinogenic role of
melatonin - potential mechanisms. Med Pr. 62:425–434. 2011.In
Polish.
|
|
29
|
Hrushesky WJ, Grutsch J, Wood P, Yang X,
Oh EY, Ansell C, Kidder S, Ferrans C, Quiton DF, Reynolds J, et al:
Circadian clock manipulation for cancer prevention and control and
the relief of cancer symptoms. Integr Cancer Ther. 8:387–397. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mao L, Yuan L, Slakey LM, Jones FE, Burow
ME and Hill SM: Inhibition of breast cancer cell invasion by
melatonin is mediated through regulation of the p38
mitogen-activated protein kinase signaling pathway. Breast Cancer
Res. 12:R1072010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mediavilla MD, Sanchez-Barcelo EJ, Tan DX,
Manchester L and Reiter RJ: Basic mechanisms involved in the
anti-cancer effects of melatonin. Curr Med Chem. 17:4462–4481.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Santoro R, Marani M, Blandino G, Muti P
and Strano S: Melatonin triggers p53Ser phosphorylation and
prevents DNA damage accumulation. Oncogene. 31:2931–2942. 2012.
View Article : Google Scholar
|
|
33
|
Schernhammer ES, Razavi P, Li TY, Qureshi
AA and Han J: Rotating night shifts and risk of skin cancer in the
nurses’ health study. J Natl Cancer Inst. 103:602–606. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fan L, Sun G, Ma T, Zhong F and Wei W:
Melatonin overcomes apoptosis resistance in human hepatocellular
carcinoma by targeting survivin and XIAP. J Pineal Res. 55:174–183.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu L, Xu Y and Reiter RJ: Melatonin
inhibits the proliferation of human osteosarcoma cell line MG-63.
Bone. 55:432–438. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Trubiani O, Recchioni R, Moroni F,
Pizzicannella J, Caputi S and Di Primio R: Melatonin provokes cell
death in human B-lymphoma cells by mitochondrial-dependent
apoptotic pathway activation. J Pineal Res. 39:425–431. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hong Y, Won J, Lee Y, Lee S, Park K, Chang
KT and Hong Y: Melatonin treatment induces interplay of apoptosis,
autophagy, and senescence in human colorectal cancer cells. J
Pineal Res. 56:264–274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jardim-Perassi BV, Arbab AS, Ferreira LC,
Borin TF, Varma NR, Iskander AS, Shankar A, Ali MM and de Campos
Zuccari DA: Effect of melatonin on tumor growth and angiogenesis in
xenograft model of breast cancer. PLoS One. 9:e853112014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jung-Hynes B, Schmit TL, Reagan-Shaw SR,
Siddiqui IA, Mukhtar H and Ahmad N: Melatonin, a novel Sirt1
inhibitor, imparts antiproliferative effects against prostate
cancer in vitro in culture and in vivo in TRAMP model. J Pineal
Res. 50:140–149. 2011.
|
|
40
|
Paroni R, Terraneo L, Bonomini F, Finati
E, Virgili E, Bianciardi P, Favero G, Fraschini F, Reiter RJ,
Rezzani R, et al: Antitumour activity of melatonin in a mouse model
of human prostate cancer: Relationship with hypoxia signalling. J
Pineal Res. 57:43–52. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rubin BP, Nishijo K, Chen HI, Yi X,
Schuetze DP, Pal R, Prajapati SI, Abraham J, Arenkiel BR, Chen QR,
et al: Evidence for an unanticipated relationship between
undifferentiated pleomorphic sarcoma and embryonal
rhabdomyosarcoma. Cancer Cell. 19:177–191. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Borenfreund E and Puerner JA: Toxicity
determined in vitro by morphological alterations and neutral red
absorption. Toxicol Lett. 24:119–124. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Repetto G, del Peso A and Zurita JL:
Neutral red uptake assay for the estimation of cell
viability/cytotoxicity. Nat Protoc. 3:1125–1131. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Battistelli M, Salucci S, Burattini S and
Falcieri E: Further considerations on in vitro skeletal muscle cell
death. Muscles Ligaments Tendons J. 3:267–274. 2013.
|
|
45
|
Salucci S, Burattini S, Battistelli M,
Baldassarri V, Curzi D, Valmori A and Falcieri E: Melatonin
prevents chemical-induced haemopoietic cell death. Int J Mol Sci.
15:6625–6640. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View Article : Google Scholar
|
|
48
|
Chipuk JE, Moldoveanu T, Llambi F, Parsons
MJ and Green DR: The BCL-2 family reunion. Mol Cell. 37:299–310.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang S, Zuo L, Gui S, Zhou Q, Wei W and
Wang Y: Induction of cell differentiation and promotion of endocan
gene expression in stomach cancer by melatonin. Mol Biol Rep.
39:2843–2849. 2012. View Article : Google Scholar
|
|
50
|
Sainz RM, Mayo JC, Tan DX, León J,
Manchester L and Reiter RJ: Melatonin reduces prostate cancer cell
growth leading to neuroendocrine differentiation via a receptor and
PKA independent mechanism. Prostate. 63:29–43. 2005. View Article : Google Scholar
|
|
51
|
Vijayalaxmi Thomas CR Jr, Reiter RJ and
Herman TS: Melatonin: From basic research to cancer treatment
clinics. J Clin Oncol. 20:2575–2601. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Panzer A and Viljoen M: The validity of
melatonin as an oncostatic agent. J Pineal Res. 22:184–202. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lissoni P, Barni S, Mandalà M, Ardizzoia
A, Paolorossi F, Vaghi M, Longarini R, Malugani F and Tancini G:
Decreased toxicity and increased efficacy of cancer chemotherapy
using the pineal hormone melatonin in metastatic solid tumour
patients with poor clinical status. Eur J Cancer. 35:1688–1692.
1999. View Article : Google Scholar
|
|
54
|
Reiter RJ, Tan DX, Sainz RM, Mayo JC and
Lopez-Burillo S: Melatonin: Reducing the toxicity and increasing
the efficacy of drugs. J Pharm Pharmacol. 54:1299–1321. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fan LL, Sun GP, Wei W, Wang ZG, Ge L, Fu
WZ and Wang H: Melatonin and doxorubicin synergistically induce
cell apoptosis in human hepatoma cell lines. World J Gastroenterol.
16:1473–1481. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Reiter RJ and Tan DX: What constitutes a
physiological concentration of melatonin? J Pineal Res. 34:79–80.
2003. View Article : Google Scholar
|
|
57
|
Venegas C, García JA, Escames G, Ortiz F,
López A, Doerrier C, García-Corzo L, López LC, Reiter RJ and
Acuña-Castroviejo D: Extrapineal melatonin: Analysis of its
subcellular distribution and daily fluctuations. J Pineal Res.
52:217–227. 2012. View Article : Google Scholar
|
|
58
|
Seabra ML, Bignotto M, Pinto LR Jr and
Tufik S: Randomized, double-blind clinical trial, controlled with
placebo, of the toxicology of chronic melatonin treatment. J Pineal
Res. 29:193–200. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Reiter RJ, Tan DX, Rosales-Corral S and
Manchester LC: The universal nature, unequal distribution and
antioxidant functions of melatonin and its derivatives. Mini Rev
Med Chem. 13:373–384. 2013.
|
|
60
|
Sainz RM, Mayo JC, Rodriguez C, Tan DX,
Lopez-Burillo S and Reiter RJ: Melatonin and cell death:
Differential actions on apoptosis in normal and cancer cells. Cell
Mol Life Sci. 60:1407–1426. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wölfler A, Caluba HC, Abuja PM, Dohr G,
Schauenstein K and Liebmann PM: Prooxidant activity of melatonin
promotes fas-induced cell death in human leukemic Jurkat cells.
FEBS Lett. 502:127–131. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Bizzarri M, Proietti S, Cucina A and
Reiter RJ: Molecular mechanisms of the pro-apoptotic actions of
melatonin in cancer: A review. Expert Opin Ther Targets.
17:1483–1496. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Galano A, Tan DX and Reiter RJ: On the
free radical scavenging activities of melatonin’s metabolites, AFMK
and AMK. J Pineal Res. 54:245–257. 2013. View Article : Google Scholar
|
|
64
|
Bejarano I, Espino J, Barriga C, Reiter
RJ, Pariente JA and Rodríguez AB: Pro-oxidant effect of melatonin
in tumour leucocytes: Relation with its cytotoxic and pro-apoptotic
effects. Basic Clin Pharmacol Toxicol. 108:14–20. 2011. View Article : Google Scholar
|
|
65
|
Sánchez-Sánchez AM, Martín V,
García-Santos G, Rodríguez-Blanco J, Casado-Zapico S,
Suarez-Garnacho S, Antolín I and Rodriguez C: Intracellular redox
state as determinant for melatonin antiproliferative vs cytotoxic
effects in cancer cells. Free Radic Res. 45:1333–1341. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Büyükavci M, Ozdemir O, Buck S, Stout M,
Ravindranath Y and Savaşan S: Melatonin cytotoxicity in human
leukemia cells: Relation with its pro-oxidant effect. Fundam Clin
Pharmacol. 20:73–79. 2006. View Article : Google Scholar : PubMed/NCBI
|