Introduction
The tumor-suppressor p53 is a potent transcription
factor that controls a major pathway protecting cells from
malignant transformation (1). As
such, it is the most frequently inactivated protein in human tumors
(2). The murine double minute 2
(MDM2) protein is one of the most well studied negative regulators
of the p53 protein (3). It is
currently known that MDM2 is overexpressed in a variety of human
tumors, which results in the inactivation of wild-type (wt)-p53
protein, with an effect similar to that of mutations in the p53
gene (4,5). Indeed, tumors with MDM2 gene
amplification almost exclusively express wt-p53 (6). Therefore, reactivating p53 activity by
antagonizing MDM2 offers a new therapeutic strategy in tumors that
retain wt-p53 (7,8).
Ribosomal protein L23 (RPL23) was discovered to be
capable of inhibiting MDM2-mediated p53 ubiquitination through
direct binding to MDM2, and subsequently induce the p53 level as
well as its activity (9,10). In addition, ectopi-cally expressed
RPL23 has been proved to be capable of interacting with MDM2 in
both the nucleus and cytoplasm, which indicates that MDM2 is
retained in the nucleus and cytoplasm as a complex, and this
complex formation represents one more mechanism by which RPL23
indirectly inhibits MDM2-p53 binding (10). Therefore, there is a rationale for
the use of RPL23 as a new target of gene therapy for tumors with
wt-p53.
Many studies have shown that human colorectal
carcinoma LoVo cells have the wt-p53 gene and the expressed p53
protein is functional and capable of inducing expression of the
target genes (11–13). Therefore, LoVo cells are a suitable
cell model with which to study the therapeutic utility of
reactivating p53 in tumors with wt-p53. In the present study, we
developed a recombinant adenoviral vector expressing the RPL23 gene
under the control of the carcinoembryonic antigen (CEA) promoter
(rAd/CEA-RPL23), and by using a LoVo cell model, we tested the
therapeutic efficacy of RPL23 gene transfer on human colorectal
carcinoma containing wt-p53 in vitro and in vivo.
Materials and methods
Cell lines and reagents
Three cell lines were used in this study, including
human colorectal cancer cell lines LoVo (wt-p53 gene) and HT29
(mutant p53 gene) (11), and normal
human skin fibroblast (HSF) cells. All cells were maintained in
RPMI-1640 supplemented with 10% FCS (Invitrogen Corp., Carlsbad,
CA, USA), at 37°C, in a humidified atmosphere containing 5%
CO2. Mouse monoclonal antibodies specific for p53
(DO-1), p21 and PUMA were obtained from Santa Cruz Biotechnology
Inc. (Santa Cruz, CA, USA), and mouse monoclonal β-actin antibody
was obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA).
The ApoAlert™ CPP32/Caspase-3 Assay kit was purchased from Clontech
Laboratories Inc. (Palo Alto, CA, USA). All reagents were used in
different concentrations as indicated.
Construction of the recombinant
adenovirus
Human CEA promoter fragment was obtained by PCR from
human genomic DNA (primers: forward, 5′-GAAGATCTAGAGCATGGGGA
GACCCG-3′ and reverse, 5′-ATTTGCGGCCGCTCTGTGG AGAAGAGCTTG-3′), and
cloned into the shuttle plasmid pAdTrack resulting in
pAdTrack-CEAp. The full-length of human RPL23 cDNA with a 6-his tag
was cut from pcDNA3.1-RPL23 constructed by our laboratory, and
subcloned into pAdTrack-CEAp resulting in pAdtrack-CEAp-RPL23 which
was linearized by PmeI and transformed into AdEasier-1
cells. Transformants were selected on LB agar plates containing 25
µg/ml kanamycin, and positive pAdEasy-CEAp-RPL23 was
identified by electrophoretic analysis of the PCR products
(primers: forward, 5′-CTGCTGGGTTTCTCTGTCACA AAG-3′ and reverse,
5′-ATGGTGATGGTGATGATGTGC AAT-3′), then digested with PacI,
and transfected into 293 cells to produce the recombinant
adenovirus Ad/CEA-RPL23 (rAd/CEA-RPL23). RT-PCR analysis was
performed to determine the specific expression of RPL23 gene
transcription driven by the CEA promoter in human colorectal cancer
LoVo and HT29 cells (primers: forward, 5′-GCCGCGATATCATGG GCATGT-3′
and reverse, 5′-GGTGATGGTGATGATGTGC AAT-3′). rAd/CEA-lacZ was
constructed by a similar method. The adenoviral transduction
efficiency of the three tested cells was evaluated by flow
cytometry (Epics XL; Coulter, Miami, FL, USA) using
adenovirus-expressing green fluorescent protein (GFP) (rAd5-GFP)
alone (14).
Assay of cell growth
Cells were plated in triplicate wells into a 96-well
plate at a density of 5×103 cells/100 µl, and
cultured routinely for 24 h. The supernatant was discarded, and
infected with recombinant adenoviruses at various multiplicity of
infection (MOI) for 24 h, and remained in culture with RPMI-1640
supplemented with 10% FCS. Four days after adenovirus infection,
cell growth was assessed by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (15).
Cell cycle analysis
LoVo cells cultured in 6-well plates (Corning) were
infected with the recombinant adenoviruses at various MOI for 48 h,
and then were harvested by centrifugation. After washing with
ice-cold PBS, the cells were suspended in ~0.5 ml of 70% alcohol
and kept at 4°C for 30 min. The suspension was filtered through
50-µm nylon mesh, and the DNA content of stained nuclei was
analyzed by a flow cytometer (Epics XL; Coulter). The cell cycle
was analyzed using Multicycle-DNA Cell Cycle Analyzed software.
Detection of apoptosis
Detection of apoptotic cells by flow cytometry was
performed as described previously (16). LoVo cells cultured in 6-well plates
(Corning) were infected with recombinant adenoviruses at various
MOI for 48 h before cells were harvested, and then Annexin
V/propidium iodide binding assay was performed by a flow cytometer
(Epics XL; Coulter).
Western blot analysis
LoVo cells infected with recombinant adenoviruses at
various MOI for 48 h were collected, washed twice in cold PBS, and
lysed at 4°C in lysis buffer using protease and phosphatase
inhibitors as described previously (15). Cell lysates containing 50 µg
total protein were resolved in 10% SDS-PAGE, transferred to
nitrocellulose polyvinylidene diflu-oride membranes (Amersham
Pharmacia Biotech, Piscataway, NJ, USA), and probed with primary
antibodies. Detection and signal visualization were conducted using
appropriate secondary antibodies conjugated with horseradish
peroxidase (Santa Cruz Biotechnology Inc.) and enhanced
chemiluminescence reagents (Pierce, Rockford, IL, USA) according to
the manufacturer’s instructions.
5-Fluorouracil (5-FU) sensitivity of
tumor cells infected with rAd/CEA-RPL23
LoVo cells were seeded in triplicate in 96-well
plates at a density of 5×103 cells/well in 100 µl
of medium. Twenty-four hours later, the cells were infected with
rAd/CEA-RPL23 (or rAd/CEA-lacZ) at an MOI of 50. After another 24 h
of incubation, the medium was changed to fresh medium containing 1
µmol/l 5-FU. The cells were then cultured for an additional
24 h before being harvested for apoptotic analysis or caspase-3
activity analysis.
Measurement of caspase-3 activity
Caspase-3 activity was determined according to the
user’s manual for the ApoAlert™ CPP32/Caspase-3 Assay kit. The
supernatant obtained by a centrifugation of lysed cells was added
to the reaction mixture containing dithiothreitol and caspase-3
substrate (N-acetyl-Asp-Glu-Val-Asp-p-nitroanilide) and incubated
for 1 h at 37°C. Absorbance of the chromophore p-nitroanilide
produced was measured at 405 nm. The standard curves were obtained
from the absorbance of p-nitroanilide standard reagent diluted with
cell lysis buffer (up to 20 nM). One unit of the enzyme was defined
as the activity producing 1 nmol of p-nitroanilide.
In vivo experiments using animal
models
Male Balb/c athymic nude mice (6- to 8-weeks of age)
(Shanghai Laboratory Animal Center of the Chinese Academy of
Sciences) were used for the experiments, as approved by the
Institutional Animal Care and Use Committee at the Southwest
Hospital. In addition, the experiments were conducted according to
guidelines for the Welfare of Animals in Experimental Neoplasia
published by The United Kingdom Coordinating Committee on Cancer
Research.
A mouse subcutaneous tumor model was established as
described previously (17). LoVo
cells (1×106 in PBS/100 µl) were injected
subcutaneously into the right flanks of nude mice. Forty days after
tumor cell inoculation, the mice were divided randomly into 3
groups (5 mice/group) and were treated every 3 days for a total of
5 times by way of multiple-center intra-tumor injection of
rAd/CEA-RPL23 or rAd/CEA-lacZ at 5×107 plaque forming
units (pfu)/50 µl per animal or PBS as a blank control.
Serial changes in tumor volume were estimated every 3 days after
the start of the recombinant adenovirus treatment. The volume of
the tumors was calculated according to the formula: Tumor volume =
(length × width2)/2.
In order to evaluate the therapeutic efficacy of
5-FU combined with rAd/CEA-RPL23 on disseminated colorectal
carcinoma, a nude mouse model was established by intraperitoneal
inoculation of 2×106 LoVo cells (18). Seven days later, the mice were
divided randomly into the following 5 groups (10 mice/group)
according to treatment schedules: a, injection of PBS as blank
control; b, injection of 5-FU alone; c, injection of rAd/CEA-RPL23
alone; d, injection of rAd/CEA-RPL23 followed by 5-FU; and e,
injection of rAd/CEA-lacZ followed by 5-FU. In groups c, d, and e,
the adenovirus (2×108 pfu/200 µl) was injected
into mice via tail vein at days 7, 10, 13, 16 and 19 after the
intraperitoneal inoculation of LoVo cells. In all groups, 5-FU (500
mg/kg body weight) or PBS was administered to mice via tail vein
injection one day after the adenovirus injection. Survival of the
animals was observed daily.
Statistical analysis
Quantitative results are expressed as the mean ± SE.
Statistical analysis was performed by using ANOVA and the LSD
t-test. The survival rates were estimated using the Kaplan-Meier
method, and the differences were analyzed by using the log-rank
test to compare the resulting curves of the treatment groups.
P<0.05 was considered as statistically significant. All
statistical analyses were performed using SPSS14.0 software (SPSS
Inc., Chicago, IL, USA).
Results
Construction of rAd/CEA-RPL23 and the
CEA-dependent RPL23 gene expression in human colorectal cancer
cells
Researchers have found that the CEA promoter
specifically promotes the exogenous gene expression in CEA-positive
tumor cells, for which it has been extensively used in the target
gene therapy of tumors (19–21).
In the present study, we obtained the CEA promoter fragment by PCR
(Fig. 1A), and using it, we
constructed a new replication-deficient recombinant adenovirus,
termed rAd/CEA-RPL23, containing the RPL23 gene under control of
the CEA promoter (Fig. 1B). Through
flow cytometry assay, the efficiency of adenovirus infection was
evaluated by counting the percentage of GFP-positive cells after
rAd-GFP infection, and as shown in Fig.
1C, among the LoVo, HT29 and HSF cells, there was no
significant difference in the infection rate of the recombinant
adenovirus serotype 5 (rAd5) when the MOI was set at 10, 50 or 100.
Furthermore, by RT-PCR analysis, we determined that upon
rAd/CEA-RPL23 infection at the same dose (50 MOI), the RPL23 gene
product was expressed specifically in the CEA-positive human
colorectal cancer LoVo and HT29 cells (17,22),
but not in the CEA-negative HSF cells (Fig. 1D).
 | Figure 1Construction and identification of
rAd/CEA-RPL23. (A) CEA promoter fragment amplified by PCR (414 bp).
Lane 1, DNA marker; lane 2, CEA promoter. (B) Target segment in
pAdEasy-CEAp-RPL23 amplified by PCR (823 bp). Lane 1, DNA marker;
lane 2, pAdEasy-CEAp-RPL23; lane 3, pAdEasy. (C) Infection rate of
recombinant adenovirus. The efficiency of adenovirus (rAd5)
infection into tested cells was evaluated through flow cytometry
assay. When the MOI was set at 50, the infection rate of the
recombinant adenovirus in the LoVo, HT29 and HSF cells was
86.3±4.9, 88.2±5.1 and 79.4±4.1%, respectively, and at MOI 100 the
rate was > 95% in all the three cell lines (n=4, P<0.05). (D)
RT-PCR products of the RPL23 gene transcripts in cells infected
with rAd/CEA-RPL23 (447 bp). Lane 1, DNA marker; lane 2, LoVo; lane
3, HT29; lane 4, HSF. CEA, carcinoembryonic antigen; RPL23,
ribosomal protein L23; MOI, multiplicity of infection; HSF, human
skin fibroblasts; rAd5, recombinant adenovirus serotype 5. |
Growth inhibitory effect of rAd/CEA-RPL23
on colorectal cancer cells
To show that the growth inhibitory effect of
rAd/CEA-RPL23 is dependent on p53 gene status in colorectal cancer
cells, wt-p53 LoVo and mutant-p53 HT29 colorectal carcinoma cell
lines were infected with increasing MOI of the recombinant
adenovirus. Through MTT assay, a dose-dependent growth inhibitory
activity of rAd/CEA-RPL23 infection was revealed in the LoVo but
not in the HT29 cells (Fig. 2A). As
depicted in Fig. 2B, compared with
control rAd/CEA-lacZ, rAd/CEA-RPL23 infection at MOI 50 and 100
decreased the growth of viable LoVo cells by 20 and 27%,
respectively. In regards to the growth of HT29 cells with mutant
p53 and CEA-negative HSF cells, rAd/CEA-RPL23 treatment did not
differ from the rAd/CEA-lacZ treatment (Fig. 2A, P>0.05).
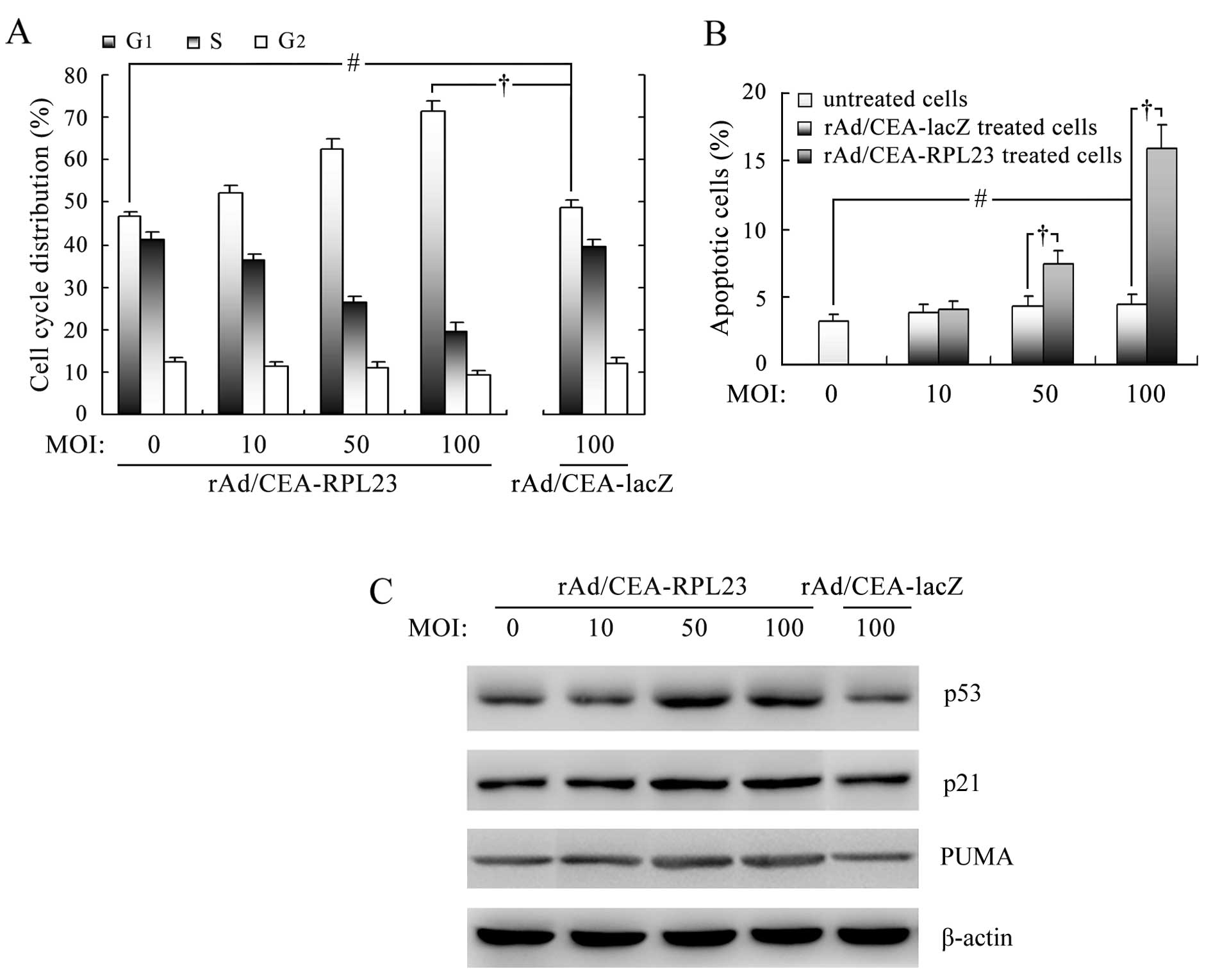
rAd/CEA-RPL23 induces cell cycle arrest
in LoVo cells through stabilization of p53 and upregulation of
p21
The above data demonstrated that rAd/CEA-RPL23
infection significantly inhibited the growth of LoVo cells in
vitro. To determine whether rAd/CEA-RPL23 induces endogenous
p53 accumulation and thus consequent cell cycle arrest in LoVo
cells, flow cytometric assay and western blot analysis were
performed. As shown in Fig. 3A, 48
h after infection with rAd/CEA-RPL23 at MOI 10, 50 and 100, the
fraction of LoVo cells in the G1 phase was increased approximately
by 5.4, 34.2 and 52.5%, respectively, associated with a
concentration-dependent decrease in the S phase fraction,
indicating cell cycle arrest at the G1-S checkpoint. As expected,
in LoVo cells infected with rAd/CEA-lacZ, no significant cell cycle
arrest was observed (P>0.05). Western blot analysis further
revealed that the level of endogenous p53 protein in LoVo cells was
increased upon rAd/CEA-RPL23 infection, indicating that
adenovirus-mediated RPL23 expression could induce stabilization and
accumulation of wt-p53 protein through inhibiting MDM2-p53
interaction (Fig. 3C). A parallel
increase in the expression of cyclin-dependent kinase inhibitor
p21, the known transcriptional target of p53 (23), was found in LoVo cells upon
rAd/CEA-RPL23 infection (Fig. 3C),
suggesting that the accumulation of wt-p53 protein induced by
rAd/CEA-RPL23 was functional and capable of mediating downstream
signal transduction.
rAd/CEA-RPL23 induces apoptosis in LoVo
cells through activation of the p53 apoptotic pathway
Since we also observed significant cell death in
LoVo cells upon rAd/CEA-RPL23 infection, we next assessed the
apoptosis using Annexin V-propidium iodide staining and flow
cytometry. As shown in Fig. 3B,
treatment of LoVo cells with rAd/CEA-RPL23 for 48 h at 50 and 100
MOI resulted in 7.5 and 16.1% apoptotic cell death, respectively
(P<0.05), and under similar conditions, rAd/CEA-lacZ did not
show the ability to induce apoptosis (P>0.05). By western blot
analysis, we further revealed that the expression level of
pro-apoptotic protein PUMA, a known transcriptional target of p53
(24), was increased in the LoVo
cells upon rAd/CEA-RPL23 infection (Fig. 3C), suggested that the accumulation
of wt-p53 protein induced by rAd/CEA-RPL23 was also capable of
triggering the p53 apoptotic pathway.
Intratumoral injection of rAd/CEA-RPL23
suppresses the growth of established LoVo subcutaneous tumors in
nude mice
To test the potential utility of RPL23 gene therapy
in the treatment of human colorectal carcinoma, we tested the
efficacy of rAd/CEA-RPL23 in vivo using a mouse subcutaneous
tumor model. One week after inoculation of LoVo cells,
rAd/CEA-RPL23, rAd/CEA-lacZ or PBS was administered locally by way
of multiple-center intratumor injection. As shown in Fig. 4, the LoVo tumor growth was
significantly suppressed in the mice given treatment of
rAd/CEA-RPL23 as compared to that in the control groups
(P<0.05). The size of the tumors between the rAd/CEA-RPL23 group
and the rAd/CEA-lacZ or PBS group was significant different from
day 20 through the experiment period (P<0.05). The difference
between the rAd/CEA-lacZ and PBS group was not statistically
significant (P>0.05).
Stabilization of wt-p53 protein upon
rAd/CEA-RPL23 infection enhances 5-FU-induced apoptosis in LoVo
cells
Research has shown that loss of p53 gene function in
colorectal carcinoma cells abolishes the apoptotic response to 5-FU
(25). In the present study,
combined treatment of LoVo cells with a relatively low dose of
rAd/CEA-RPL23 and 5-FU was performed to evaluate whether
stabilization of wt-p53 protein upon rAd/CEA-RPL23 infection could
enhance 5-FU-induced apoptosis. Although single agent 5-FU at 1
µmol/l was able to induce apoptosis in LoVo cells, the
apoptosis induced by treatment in combination with rAd/CEA-RPL23 at
MOI 50 was significantly greater than that following treatment of
5-FU alone. As shown in Fig. 5A,
the apoptotic rate in the 5-FU alone group and rAd/CEA-RPL23 alone
group was 6.0 and 7.5%, respectively, while, the apoptotic rate in
the combined treatment group was 25.3%, which was more than the
additive effect of each treatment separately (25.3% vs. 6.0+7.5%).
These results were confirmed by caspase-3 activity assay. Caspase-3
is a central executioner of apoptosis (26). In the present study, it was found
that the caspase-3 activity of LoVo cells treated with the
combination of 50 MOI rAd/CEA-RPL23 and 1 µmol/l 5-FU was
increased significantly to 5.78-fold when compared with the
control, whereas the activity following treatment with single 5-FU
or rAd/CEA-RPL23 was only increased slightly to 1.64- and 1.91-fold
of the control, respectively (Fig.
5B). Western blot analysis gave similar result, which showed
that upon combination treatment with 5-FU and rAd/CEA-RPL23, the
expression level of cleaved caspase-3 was markedly increased as
compared to the levels following treatment of 5-FU or rAd/CEA-RPL23
alone (Fig. 5C).
rAd/CEA-RPL23 improves the therapeutic
efficacy of 5-FU in a murine model with disseminated LoVo
tumors
To evaluate the therapeutic efficacy of 5-FU
combined with rAd/CEA-RPL23 in vivo, a murine model with
intraperitone-ally disseminated LoVo tumors that closely resemble
human advanced colorectal carcinoma was designed. Fig. 6 shows the survival rates of animals
after the inoculation of LoVo cells. The median survival time of
the mice treated with 5-FU combined with rAd/CEA-RPL23 (39±3.3
days) was longer than that of the mice treated with 5-FU alone
(25±3.8 days) or 5-FU combined with rAd/CEA-lacZ (23±3.0 days).
Log-rank test revealed that the survival was significantly longer
for mice in the 5-FU+rAd/CEA-RPL23 group as compared with that in
the 5-FU group or 5-FU+rAd/CEA-lacZ group (P<0.05).
Additionally, treatment of rAd/CEA-RPL23 alone also showed a
detectable therapeutic effect on the survival of mice as compared
with the blank control (19±1.3 days vs. 15±2.0 days), although the
difference was not statistically significant (P>0.05).
 | Figure 6Therapeutic effect of 5-FU combined
with rAd/CEA-RPL23 on survival periods in a disseminated colorectal
carcinoma model. The nude mice which were intraperitoneally
inoculated with LoVo cells (2×106 cells/mouse) were
randomly divided into the following 5 groups (10 mice/group)
according to treatment schedules: group a, ◆, injection of PBS as
blank control; b, ○, injection of rAd/CEA-RPL23 alone; c, •,
injection of 5-FU alone; d, □, injection of rAd/CEA-lacZ followed
by 5-FU; e, ■, injection of rAd/CEA-RPL23 followed by 5-FU.
Survival of animals was observed everyday. 5-FU, 5-fluorouracil;
CEA, carcinoembryonic antigen; RPL23, ribosomal protein L23. |
Discussion
Since overexpression or hyperactivation of MDM2
contributes to functional inactivation of wt-p53 in nearly 50% of
tumors (27), and since MDM2 is
capable of attenuating the effectiveness of chemotherapies that
induce p53 (28), antagonizing MDM2
is being considered an attractive strategy for the treatment of
tumors (5,7). Different approaches have been
exploited in this strategy including inhibition of MDM2 expression,
MDM2 ubiquitin ligase activity and MDM2-p53 binding (7). Recently, a small-molecule compound
termed Nutlin-3a, which specifically inhibited the MDM2-p53
interaction and thus led to stabilization of p53 and activation of
the p53 pathway, was discovered and has been reported to display a
potent growth-inhibitory effect on a variety of tumor cells that
contain wt-p53 (7,29). The p14ARF protein,
encoded by the INK4a/ARF locus, was found to bind to MDM2 directly
and inhibit the ubiquitin E3 ligase function of MDM2. It has been
reported that the p14ARF gene transfer mediated by
adenovirus vector inhibited the growth and enhanced the response of
chemotherapies in a variety of wt-p53 tumor cells (30–32).
RPL23 is another cellular protein with the capacity of binding to
MDM2 and inhibiting the ubiquitin E3 ligase function of MDM2
(9,10), and Jin et al reported that
the ability of RPL23 to inhibit MDM2’s E3 ligase function was
comparable to that of p14ARF (9), suggesting a rationale for using RPL23
as a new target for tumor gene therapy.
The therapeutic utility of reactivating p53 depends
on two critical factors: defective p53 signaling in tumor cells
with wt-p53 may attenuate or disable the therapeutic response; and
possible growth suppressive and/or apoptotic activity of p53 in
normal tissues may narrow or eliminate the therapeutic window
(7). Human colorectal carcinoma
LoVo cells have been confirmed to express functional p53 protein
(11–13). Therefore, in the present study, LoVo
cells were used as a model system to evaluate the therapeutic
efficacy of RPL23 gene transfer on human tumors with wt-p53. The
other key factor regarding the therapeutic utility of
pharmacological p53 activation is p53 toxicity to normal tissues.
Since embryonic genes such as CEA are usually transcriptionally
silent in adult normal tissues but overexpressed in malignant
tumor, the use of an embryonic gene promoter to regulate expression
of a target gene for the purpose of reducing the toxicity of gene
transfer into normal tissues is popular (19–21).
In the present study, we constructed a recombinant adenoviral
vector expressing the RPL23 gene driven by the CEA promoter
(rAd/CEA-RPL23). In theory, rAd/CEA-RPL23 can deliver the RPL23
gene specifically to colorectal cancer cells but not to normal
tissue cells, by which, rAd/CEA-RPL23 infection is expected to be
slightly toxic to normal tissues.
Through flow cytometry and western blot analysis, we
validated that the exogenous RPL23 protein expression by adenoviral
vector under control of the CEA promoter was restricted to
CEA-positive LoVo and HT29 human colorectal carcinoma cells but not
CEA-negative HSF cells. Furthermore, we determined that the
exogenous RPL23 protein could stabi-lize endogenous p53 protein and
thus induce cell cycle arrest and apoptosis in wt-p53 LoVo cells.
An in vivo study showed that the growth of subcutaneous
tumors derived from LoVo cells was significantly inhibited by
direct intratumor injection of rAd/CEA-RPL23. Our study also
discovered that rAd/CEA-RPL23 infection had no obvious effect on
the growth of p53-mutant HT29 human colorectal cancer cells, which
suggested that the growth inhibiting effect of exogenous RPL23
expression was mainly via p53-dependent mechanisms.
5-FU is a widely used chemotherapeutic drug for the
treatment of human colorectal carcinoma. The antitumor activity of
5-FU has been attributed in part to its ability to induce
p53-dependent cell growth arrest and apoptosis (33,34).
Since hyperactivation of MDM2 has been proved to be capable of
attenuating 5-FU-induced toxicity by inhibiting p53 activation
(35), we reasoned that a
combination treatment of 5-FU and RPL23 gene transfer may improve
the prognosis of patients with colorectal carcinoma after surgery.
The results of our in vitro experiment showed that
rAd/CEA-RPL23 pretreatment in LoVo cells enhanced 5-FU-induced
apoptosis, and assay of the in vivo therapeutic efficacy
revealed that the combination treatment increased the overall
survival of mice with disseminated LoVo tumors. These results
indicate that RPL23 gene transfer may be used as part of a combined
therapy for the treatment of human tumors. Notably, the combination
treatment also showed a detectable enhancement on the survival of
mice with disseminated HT29 tumors (data not shown), indicating
that RPL23 gene transfer may influence the sensitivity of tumor
cells to antitumor drugs via a p53-independent manner. As it has
been determined that ectopically expressed RPL23 is capable of
retaining MDM2 both in the cytoplasm and the nucleus (10), we hypothesized that the exogenous
RPL23 expression may improve 5-FU efficacy in tumors with mutant
p53 via its function of blocking MDM2 nuclear translocation. For
example, MDM2 is essential to the activation of NF-κB in some tumor
cells (36,37), and therefore, the blocking of MDM2
nuclear translocation by ectopically expressed RPL23 may lead to a
reduction in NF-κB activation, which has been proved to directly
contribute to chemotherapy resistance and metastasis in human
tumors (38). Therefore,
suppression of the NF-κB pathway may be another mechanism by which
RPL23 gene transfer exhibits a synergistic effect with antitumor
drugs such as 5-FU, regardless of the p53 status.
In conclusion, the present study demonstrated that
exogenous RPL23 expression driven by the CEA promoter may be of
therapeutic value against human colorectal carcinoma that retains
wt-p53.
Acknowledgments
The present study was supported by the China
Postdoctoral Science Foundation (no. 20100481468).
References
|
1
|
Selivanova G: Wild type p53 reactivation:
from lab bench to clinic. FEBS Lett. 588:2628–2638. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khoo KH, Verma CS and Lane DP: Drugging
the p53 pathway: understanding the route to clinical efficacy. Nat
Rev Drug Discov. 13:217–236. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kruse JP and Gu W: Modes of p53
regulation. Cell. 137:609–622. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Freedman DA, Wu L and Levine AJ: Functions
of the MDM2 oncoprotein. Cell Mol Life Sci. 55:96–107. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang W and El-Deiry WS: Restoration of p53
to limit tumor growth. Curr Opin Oncol. 20:90–96. 2008. View Article : Google Scholar
|
|
6
|
Momand J, Jung D, Wilczynski S and Niland
J: The MDM2 gene amplification database. Nucleic Acids Res.
26:3453–3459. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao Y, Aguilar A, Bernard D and Wang S:
Small-molecule inhibitors of the MDM2-p53 protein-protein
interaction (MDM2 inhibitors) in clinical trials for cancer
treatment. J Med Chem. 58:1038–1052. 2015. View Article : Google Scholar
|
|
8
|
Lu C and El-Deiry WS: Targeting p53 for
enhanced radio- and chemo-sensitivity. Apoptosis. 14:597–606. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dai MS, Zeng SX, Jin Y, Sun XX, David L
and Lu H: Ribosomal protein L23 activates p53 by inhibiting MDM2
function in response to ribosomal perturbation but not to
translation inhibition. Mol Cell Biol. 24:7654–7668. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jin A, Itahana K, O’Keefe K and Zhang Y:
Inhibition of HDM2 and activation of p53 by ribosomal protein L23.
Mol Cell Biol. 24:7669–7680. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pocard M, Chevillard S, Villaudy J, Poupon
MF, Dutrillaux B and Remvikos Y: Different p53 mutations produce
distinct effects on the ability of colon carcinoma cells to become
blocked at the G1/S boundary after irradiation. Oncogene.
12:875–882. 1996.PubMed/NCBI
|
|
12
|
Shi MD, Lin HH, Lee YC, Chao JK, Lin RA
and Chen JH: Inhibition of cell-cycle progression in human
colorectal carcinoma Lovo cells by andrographolide. Chem Biol
Interact. 174:201–210. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang NH, Song LB, Wu XJ, Li RP, Zeng MS,
Zhu XF, Wan DS, Liu Q, Zeng YX and Zhang XS: Proteasome inhibitor
MG-132 modifies coxsackie and adenovirus receptor expression in
colon cancer cell line Lovo. Cell Cycle. 7:925–933. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim HJ, Cho HI, Han YH, Park SY, Kim DW,
Lee DG, Kim JH, Shin WS, Paik SY, Kim CC, et al: Efficient
transduction with recombinant adenovirus in EBV-transformed B
lymphoblastoid cell lines. J Biochem Mol Biol. 37:376–382. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Shi Y, Li X, Du R, Luo G, Xia L,
Du W, Chen B, Zhai H, Wu K, et al: Proteasome inhibitor MG132
reverses multidrug resistance of gastric cancer through enhancing
apoptosis and inhibiting P-gp. Cancer Biol Ther. 7:540–546. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li X, Zhang Y, Xiong C, Jin H, Jing B,
Zhang Y and Fan D: Overexpression of a new gene P28GANK confers
multidrug resistance of gastric cancer cells. Cancer Invest.
27:129–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Y, Chen Y, Dilley J, Arroyo T, Ko D,
Working P and Yu DC: Carcinoembryonic antigen-producing
cell-specific oncolytic adenovirus, OV798, for colorectal cancer
therapy. Mol Cancer Ther. 2:1003–1009. 2003.PubMed/NCBI
|
|
18
|
Cao G, Kuriyama S, Gao J, Kikukawa M, Cui
L, Nakatani T, Zhang X, Tsujinoue H, Pan X, Fukui H, et al:
Effective and safe gene therapy for colorectal carcinoma using the
cytosine deaminase gene directed by the carcinoembryonic antigen
promoter. Gene Ther. 6:83–90. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schrewe H, Thompson J, Bona M, Hefta LJ,
Maruya A, Hassauer M, Shively JE, von Kleist S and Zimmermann W:
Cloning of the complete gene for carcinoembryonic antigen: analysis
of its promoter indicates a region conveying cell type-specific
expression. Mol Cell Biol. 10:2738–2748. 1990.PubMed/NCBI
|
|
20
|
Qiao J, Doubrovin M, Sauter BV, Huang Y,
Guo ZS, Balatoni J, Akhurst T, Blasberg RG, Tjuvajev JG, Chen SH,
et al: Tumor-specific transcriptional targeting of suicide gene
therapy. Gene Ther. 9:168–175. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ueda K, Iwahashi M, Nakamori M, Nakamura
M, Matsuura I, Ojima T and Yamaue H: Improvement of
carcinoembryonic antigen-specific prodrug gene therapy for
experimental colon cancer. Surgery. 133:309–317. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dabrowska A, Szary J, Kowalczuk M, Szala S
and Ugorski M: CEA-negative glioblastoma and melanoma cells are
sensitive to cytosine deaminase/5-fluorocytosine therapy directed
by the carcinoembryonic antigen promoter. Acta Biochim Pol.
51:723–732. 2004.PubMed/NCBI
|
|
23
|
Chen X, Bargonetti J and Prives C: p53,
through p21 (WAF1/CIP1), induces cyclin D1 synthesis. Cancer Res.
55:4257–4263. 1995.PubMed/NCBI
|
|
24
|
Yu J and Zhang L: No PUMA, no death:
implications for p53-dependent apoptosis. Cancer Cell. 4:248–249.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sobrero A, Kerr D, Glimelius B, Van Cutsem
E, Milano G, Pritchard DM, Rougier P and Aapro M: New directions in
the treatment of colorectal cancer: a look to the future. Eur J
Cancer. 36:559–566. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kuribayashi K, Mayes PA and El-Deiry WS:
What are caspases 3 and 7 doing upstream of the mitochondria?
Cancer Biol Ther. 5:763–765. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rayburn E, Zhang R, He J and Wang H: MDM2
and human malignancies: expression, clinical pathology, prognostic
markers, and implications for chemotherapy. Curr Cancer Drug
Targets. 5:27–41. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shangary S and Wang S: Small-molecule
inhibitors of the MDM2-p53 protein-protein interaction to
reactivate p53 function: a novel approach for cancer therapy. Annu
Rev Pharmacol Toxicol. 49:223–241. 2009. View Article : Google Scholar :
|
|
30
|
Wang W, Rastinejad F and El-Deiry WS:
Restoring p53-dependent tumor suppression. Cancer Biol Ther.
2(Suppl 1): S55–S63. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Deng X, Kim M, Vandier D, Jung YJ,
Rikiyama T, Sgagias MK, Goldsmith M and Cowan KH: Recombinant
adenovirus-mediated p14(ARF) overexpression sensitizes human breast
cancer cells to cisplatin. Biochem Biophys Res Commun. 296:792–798.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hemmati PG, Gillissen B, von Haefen C,
Wendt J, Stärck L, Güner D, Dörken B and Daniel PT:
Adenovirus-mediated overexpression of p14(ARF) induces p53 and
Bax-independent apoptosis. Oncogene. 21:3149–3161. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kaeser MD, Pebernard S and Iggo RD:
Regulation of p53 stability and function in HCT116 colon cancer
cells. J Biol Chem. 279:7598–7605. 2004. View Article : Google Scholar
|
|
34
|
Sun XX, Dai MS and Lu H: 5-fluorouracil
activation of p53 involves an MDM2-ribosomal protein interaction. J
Biol Chem. 282:8052–8059. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang H, Oliver P, Zhang Z, Agrawal S and
Zhang R: Chemo-sensitization and radiosensitization of human cancer
by antisense anti-MDM2 oligonucleotides: in vitro and in vivo
activities and mechanisms. Ann NY Acad Sci. 1002:217–235. 2003.
View Article : Google Scholar
|
|
36
|
Gu L, Findley HW and Zhou M: MDM2 induces
NF-kappaB/p65 expression transcriptionally through Sp1-binding
sites: a novel, p53-independent role of MDM2 in doxorubicin
resistance in acute lymphoblastic leukemia. Blood. 99:3367–3375.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cheney MD, McKenzie PP, Volk EL, Fan L and
Harris LC: MDM2 displays differential activities dependent upon the
activation status of NFkappaB. Cancer Biol Ther. 7:38–44. 2008.
View Article : Google Scholar
|
|
38
|
Shen HM and Tergaonkar V: NFkappaB
signaling in carcinogenesis and as a potential molecular target for
cancer therapy. Apoptosis. 14:348–363. 2009. View Article : Google Scholar : PubMed/NCBI
|