Introduction
Gastric cancer (GC) remains one of the most common
causes of cancer-related death worldwide, although its incidence is
decreasing. Despite recent improvements in the early diagnosis and
effective treatment of GC, its progression and metastasis are major
contributors to GC-related death (1-3).
Therefore, an understanding of the molecular and biological changes
underlying the progression and metastasis of GC is required to
predict outcomes, personalize treatment and improve the survival
rates of GC patients.
The B-cell leukemia/lymphoma-2 (Bcl-2) protein
family regulates the integrity of the outer mitochondrial membrane
and intrinsic pathways of apoptosis. The Bcl-2 family comprises
pro- and anti-apoptotic members. The pro-apoptotic members control
the release of cytochrome c, and subsequent activation of
caspases. In contrast, anti-apoptotic members such as Bcl-2,
Bcl-xL, Bcl-w, A1 and myeloid cell leukemia-1 (Mcl-1) promote cell
survival by inhibiting pro-apoptotic proteins, including Bim, Bax,
and Bak (4–7).
Mcl-1 is a rapidly inducible, anti-apoptotic Bcl-2
protein with a very short half-life. Cells with increased Mcl-1
expression levels exhibit inhibition of apoptosis and cell cycle
progression, and chemoresistance (8–11).
Increased expression of Mcl-1 occurs in a variety of human cancers
and is strongly associated with resistance to therapies, tumor
progression, and poor prognosis in most cancers, including GC
(12–17). Therefore, Mcl-1 could be a promising
molecular target with respect to improving treatment strategies and
outcomes for cancer patients.
Epithelial-mesenchymal transition (EMT) is a complex
process that has been observed in embryonic development,
differentiation of normal tissues and organs, wound healing, and
cancer progression. During EMT, cells lose their epithelial
characteristics and gain mesenchymal phenotypes, which are
correlated with increased motility and invasion (18-22).
Mesenchymal cells tend to dedifferentiate and acquire stem cell or
tumorigenic phenotypes, such as invasion, metastasis, resistance to
apoptosis and drug resistance during EMT progression (18–22).
EMT has been implicated in cancer progression and
metastasis, and is associated with poor clinical prognosis in a
variety of human cancers (18–22).
However, the interaction between Mcl-1 and EMT in human GC is
unclear. We investigated the impact of Mcl-1 expression levels on
EMT and the underlying signaling pathways in human GC cells.
Materials and methods
Cell culture and transfection with small
interfering RNAs (siRNAs)
Human GC cell lines, AGS and SNU638, were obtained
from the American Type Culture Collection (ATCC; Manassas, VA, USA)
and the Korean Cell Line Bank (Seoul, Korea), respectively. Cells
were cultured in RPMI-1640 medium containing 25 mM HEPES and
supplemented with 10% fetal bovine serum (FBS) (both from Hyclone,
Logan, UT, USA), 50 U/ml penicillin, and 50 μg/ml
streptomycin (Gibco, Grand Island, NY, USA). Cultures were
incubated at 37°C in 5% CO2 in a humidified environment.
Cells were seeded on plates at a density such that they would be
40-50% confluent at the time of transfection. The Mcl-1-specific
and control-scrambled siRNA duplexes were purchased from Bioneer
(Daeheon, Korea) and Qiagen (Germantown, MD, USA), respectively.
The siRNAs were transfected into cells using
Lipofectamine® RNAiMAX (Invitrogen, Carlsbad, CA, USA)
according to the manufacturer’s instructions.
Reverse transcription-polymerase chain
reaction (RT-PCR) assays
Total RNA was isolated from the cells using TRIzol
reagent (Invitrogen) according to the manufacturer’s instructions.
For each sample, 1 μg of total RNA was used to generate
complementary DNA in a reaction containing 50 ng/μl oligo-dT
(Promega, Madison, WI, USA) that was incubated at 72°C for 10 min.
We then added MMLV transcription reagents (Promega) and RNAsin
(Takara, Otsu, Shiga, Japan) to each reaction and incubated the
samples at 42°C for 1 h and 72°C for 15 min. PCR amplification was
performed using gene-specific primers and GoTaq® DNA
polymerase (Promega). The primers we used were specific for Mcl-1
(5′-TCC TCT TGC CAC TTG CTT TT-3′ and 5′-TGC TGG AGT AGG AGC TGG
TT-3′); and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 5′-ACC
ACA GTC CAT GCC ATC AC-3′ and 5′-TCC ACC ACC CTG TTG CTG TA-3′).
Amplicons were separated by electrophoresis on 1% (w/v) agarose
gels containing ethidium bromide.
Western blotting
Proteins were extracted from the cells using RIPA
buffer (1 M Tris-HCl, 150 mM NaCl, 1% Triton X-100 and 2 mM EDTA)
supplemented with 1 mM PMSF, Halt™ Phosphatase Inhibitor Cocktail
and Halt™ Protease Inhibitor Cocktail (both from Thermo, Rockford,
IL, USA). Proteins were resolved by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to
polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica,
MA, USA). After blocking with 5% bovine serum albumin (BSA) buffer,
PVDF membranes were probed with the appropriate primary antibody.
We used antibodies against human Mcl-1, Snail, vimentin,
E-cadherin, phosphorylated β-catenin, β-catenin, MEK1/2,
phosphorylated ERK1/2, ERK1/2, p38 and phosphorylated p38 (all from
Cell Signaling Technology, Danvers, MA, USA). Antibodies against
human MMP-2, MMP-9, GAPDH and β-tubulin were purchased from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against human
CD44 and CD133 were purchased from R&D Systems (Minneapolis,
MN, USA) and eBioscience (San Diego, CA, USA), respectively.
Protein bands were detected using a chemiluminescent horseradish
peroxidase substrate (Millipore) and an ImageQuant™ LAS 4000
Luminescence imager (Fujifilm, Tokyo, Japan). The density ratio (%)
of protein bands was quantified using MultiGauge V3.2 image
analyzer software (Fujifilm).
Cell adhesion assays
Cell adhesion assay was conducted by coating
fibronectin (2 μg/ml; Calbiochem, La Jolla, CA, USA) and
collagen type I and IV (40 μg/ml; Corning Inc., Corning, NY,
USA), respectively. The coated wells were washed with PBS, and
blocked with 0.2% BSA medium for 30 min. Then the transfected cell
suspension was added into the coated wells and incubated for 1 h at
37°C. Non-adherent cells were removed by washing with PBS. The
attached cells were reacted with WST-1 solution (Daeil Lab Inc.,
Seoul, Korea) medium at 37°C for 1 h. The optical density was
measured at 450 nm. All experiments were carried out in
triplicate.
Cell invasion assays
We conducted cell invasion assays using Transwell
chambers with 8-μm pores (Corning Inc.). Transwell chambers
were coated with 1% gelatin in RPMI-1640 overnight and then allowed
to dry at room temperature. Cells transfected with the Mcl-1 and
scrambled siRNAs were resuspended in 120 μl of 0.2% (w/v)
BSA solution and seeded into the upper chambers. The lower chambers
contained 400 μl of 0.2% (w/v) BSA solution supplemented
with 10 μg/ml human plasma fibronectin (Calbiochem) as a
chemoattractant. After incubation for 24 h, the cells that had
migrated to the bottom surface of the Transwell were fixed with 70%
ethanol and stained with Diff-Quik solution (Sysmex, Kobe, Japan).
The cells in the upper chambers were removed using a cotton tip.
Stained cells in the lower chambers were counted using light
microscopy, from five randomly selected fields of view (0.5×0.5
mm2).
Cell migration assays
Cell migration was measured using Ibidi
Culture-Inserts (Ibidi, Regensburg, Germany). The cells transfected
with the Mcl-1 and scrambled siRNAs were seeded on the
culture-inserts and incubated at 37°C in a humidified environment.
After 24 h, the culture-inserts were gently removed using sterile
tweezers to create a cell-free gap. Cell migration into the
cell-free gap was followed for 24 h, and photographed using an
inverted microscope. The distance between gaps was normalized to 1
cm after images were captured at three random sites.
Statistical analysis
The associations between experimental groups were
analyzed using a Student’s t-test. A value of P<0.05 was
considered to indicate a statistically significant result.
Results
Impact of Mcl-1 knockdown on EMT in the
human GC cells
To study the biological role of Mcl-1 in GC
progression, we used siRNAs to knock down endogenous Mcl-1
expression in AGS and SNU638 cells. Expression levels of Mcl-1 mRNA
and protein in all tested cells were reduced following transfection
with the Mcl-1 siRNAs (Fig. 1). To
investigate the relationship between Mcl-1 and EMT in the human GC
cells, cell adhesion assays were performed. The cell adhesion
ability was measured after transfection of the siRNAs using three
cell adhesion substrates including fibronectin and collagen I and
IV. The cell adhesion to fibronectin and collagen I was
significantly increased in the Mcl-1 siRNA-transfected AGS (P=0.023
and 0.034, respectively) and SNU638 cells (P=0.045 and 0.025,
respectively) compared to that of the scrambled siRNA-transfected
cells (Fig. 2). To investigate
phenotypic changes induced by EMT, the expression levels of
EMT-associated genes (MMP-2, MMP-9, Snail, E-cadherin, and
vimentin) were also assessed. We observed lower expression levels
of vimentin, MMP-2, MMP-9 and Snail in the Mcl-1 siRNA-transfected
AGS and SNU638 cells, compared to these levels in the scrambled
siRNA-transfected cells. The E-cadherin expression level was
increased in the Mcl-1 siRNA-transfected AGS cells, but this level
was not significantly different in the SNU638 cells (Fig. 3). We investigated the possible
effect of Mcl-1 on the expression of cancer stemness markers such
as CD44 and CD133. CD44 and CD133 expression levels were unaltered
by knockdown of Mcl-1 (Fig. 3). Our
results indicate that Mcl-1 expression is associated with the
induction of molecular and cellular alterations consistent with
EMT.
 | Figure 2Mcl-1 knockdown leads to increased
cellular adhesion to fibronectin and collagen I in human GC cells.
The cell adhesion ability was measured after transfection of the
siRNAs using three cell adhesion substrates including fibronectin
and collagen I and IV. The adherent cells were stained with crystal
violet, dissolved with sodium dodecyl sulfate, and then quantified
by reading the absorbance at 540 nm using a plate reader. The cell
adhesion to fibronectin and collagen I was significantly increased
in the Mcl-1 siRNA-transfected AGS (P=0.023 and 0.034,
respectively) and SNU638 cells (P=0.045 and 0.025, respectively)
compared to the ability of the scrambled siRNA-transfected cells.
Each bar represents the mean ± SE of 3 experiments.
*P<0.05 vs. scrambled siRNA-transfected cells. SS,
scrambled siRNA; MS, Mcl-1 siRNA; FN, fibronectin; Col I, collagen
I; Col IV, collagen IV; Mcl-1, myeloid cell leukemia-1; GC, gastric
cancer. |
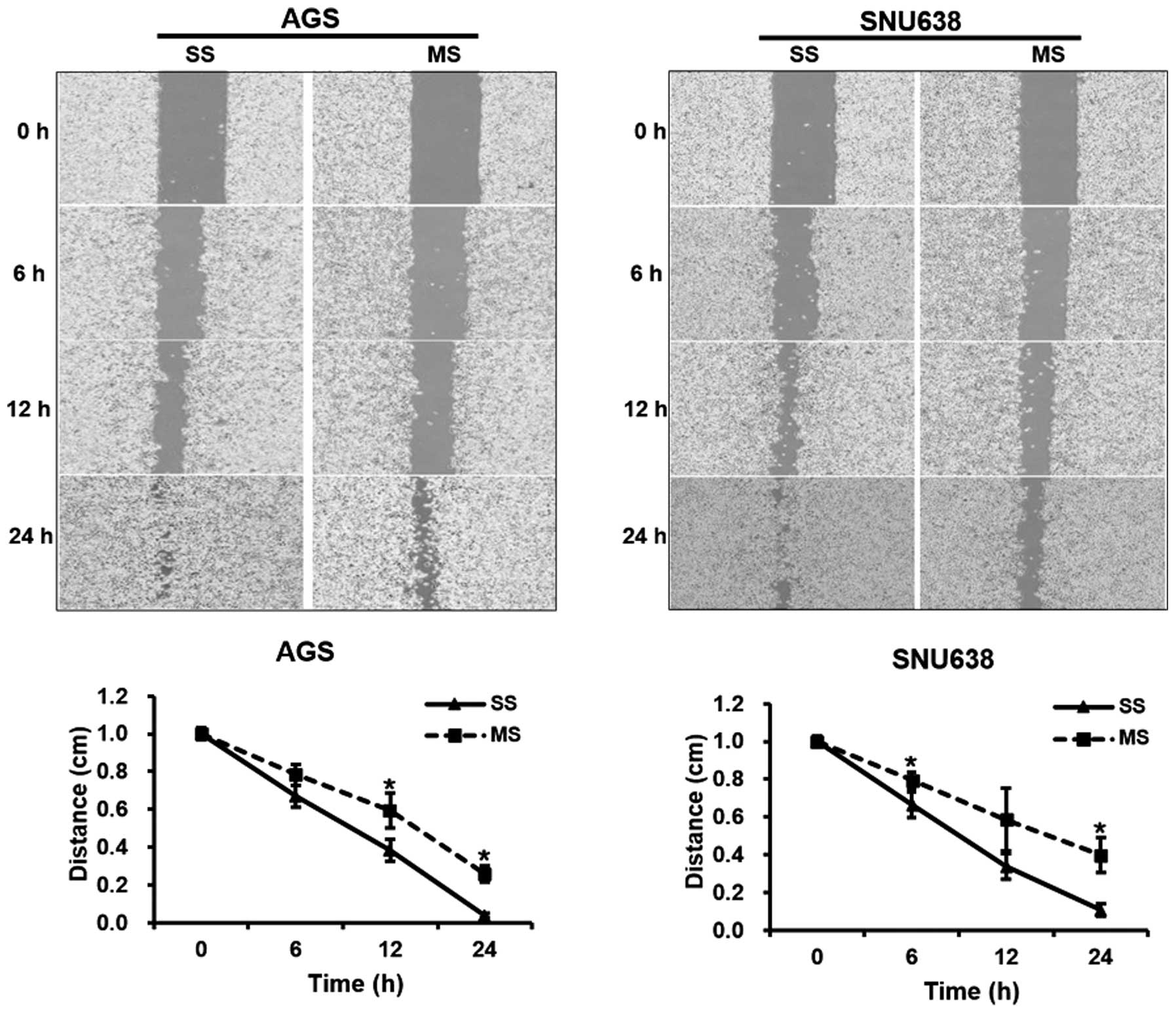
Mcl-1 knockdown affects migration and
invasion of human GC cells
For the cell migration assays, the artificial wound
gap became significantly narrower for cells transfected with the
control-scrambled siRNAs in comparison with that for the Mcl-1
siRNA-transfected cells at 12 and 24 h in the AGS cell line
(P=0.001 and 0.019, respectively). Similar results were noted at 6
and 24 h for the SNU638 cell cultures (P=0.033 and 0.023,
respectively, Fig. 4). For the cell
invasion assays, 160.3±93.8 and 117.7±70.7 invading Mcl-1
siRNA-transfected AGS and SNU638 cells, respectively, were
observed. In contrast, for cultures transfected with the scrambled
siRNAs, 424.0±146.2 and 382.7±109.4 invading AGS and SNU638 cells,
respectively were observed. These differences in invading cell
numbers were significantly different (P=0.018 for AGS cells and
P=0.009 for SNU638 cells, Fig. 5).
Our findings indicate that Mcl-1 expression is required for GC cell
migration and invasion, subsequently leading to tumor
metastasis.
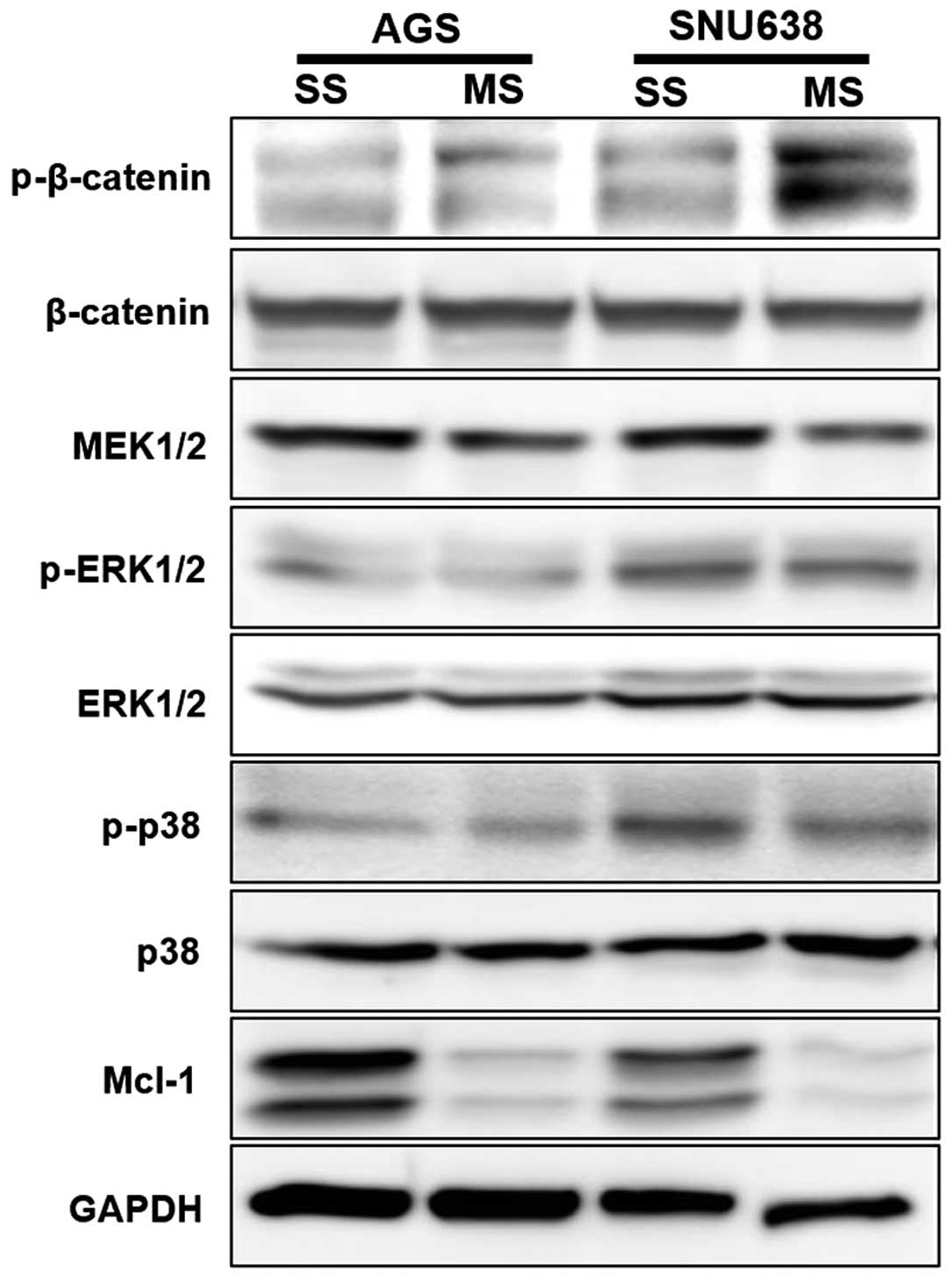
Mcl-1 knockdown affects β-catenin, MEK1/2
and MAPK signaling pathways in human GC cells
We assessed phos-phorylation levels of proteins in
the β-catenin, MEK1/2 and MAPK signaling cascades using western
blotting to determine their involvement in EMT regulation. The
phosphorylation level of β-catenin was increased in the AGS and
SNU638 cells when Mcl-1 was knocked down. Phosphorylation levels of
MEK1/2 were decreased in the AGS and SNU638 cells when Mcl-1
expression was knocked down. Phosphorylation of ERK1/2 and p38 was
decreased in the Mcl-1 siRNA-transfected SNU638 cells (Fig. 6).
Discussion
Metastatic gastric cancer (GC) is incurable and
ultimately claims the life of the majority of these patients
(1–3). Tumor metastasis is a complex process
involving tumor cells migrating from the primary tumor mass to
distant organs or tissues. The tumor microenvironment is thought to
drive tumor initiation and progression, with anti-apoptotic effects
stimulated, cell proliferation, angiogenesis, invasion, metastasis
and EMT of tumor cells observed (23,24).
EMT is a physiological process that is activated
during wound healing, inflammation or embryogenesis. Recently, EMT
has also been described for cancer cells, allowing them to acquire
motility and invasiveness. EMT is considered an essential step in
driving the early phases of tumor metastasis (18–22).
EMT induces phenotypic changes with respect to the shape and
polarity of epithelial cells. These phenotypic changes in
epithelial cells include a remodeled cytoskeleton, loss of
cell-cell adhesion, the ability to overcome anoikis and
apoptosis, and the acquisition of mobile and invasive
characteristics, which are all typical of mesenchymal cells
(18–22). Therefore, markers involved in EMT
activation may be associated with the modulation of pro- and
anti-apoptotic genes.
Mcl-1 is an anti-apoptotic Bcl-2 protein that is
highly expressed in a variety of human cancers. Expression of Mcl-1
has been shown to contribute to tumorigenesis, and is associated
with the acquisition of invasive and metastatic capabilities by
tumor cells through the inhibition of apoptosis, cell cycle
progression, promotion of cancer cell replication, invasion and
metastasis (8–11). Furthermore, expression of Mcl-1 is
associated with advanced stages and poor clinical outcome of many
human cancers including GC (12–17).
During EMT, expression of epithelial markers such as
E-cadherin, γ-catenin, cytokeratin and occludin are down-regulated
in cancer cells. Simultaneously, expression levels of mesenchymal
markers such as vimentin, fibronectin, N-cadherin, Twist and Snail
are increased. In addition, proteolytic enzymes such as MMPs, which
are required for the degradation of the extracellular matrix (ECM)
in normal tissue surrounding tumors, are activated (18–22).
These morphological and cellular alterations are critical steps in
EMT, and common steps in tumor metastasis.
First, to further explore the role of Mcl-1 in
cell-cell adhesion of human GC cells, we used three common ECM
proteins, including fibronectin and collagen I and IV, to examine
whether knockdown of Mcl-1 could affect the adhesive capacity of
human GC cells. Our study showed that knockdown of Mcl-1 led to
increased adhesion of human GC cells to fibronectin and collagen I,
but not collagen IV. This result indicates that altered expression
of Mcl-1 may be associated with altered adhesion to specific
components of the ECM such as fibronectin and collagen I in human
GC cells.
Next, we evaluated the expression of EMT-associated
genes and their corresponding proteins in human GC cells.
Expression levels of vimentin, MMP-2, MMP-9 and Snail were
decreased in cells where Mcl-1 expression was knocked down.
E-cadherin expression was increased in AGS cells following
knockdown of Mcl-1. Our results indicate a positive relationship
between Mcl-1 expression and induction of EMT in human GC
cells.
Cancer stem cells are a small subset of tumor cells
that possess extensive proliferative potential; therefore they can
initiate and propagate tumors. During EMT, epithelial cells acquire
stem cell phenotypes. There is a link between EMT and cancer stem
cells, with a correlation observed for EMT occurrence, GC
progression and resistance to treatment (25–27).
However, the expression of CD44 and CD133 was not altered by Mcl-1
knockdown in our study.
Molecular signaling pathways involved in the
induction of EMT have been identified during development,
differentiation, and carcinogenesis. Signaling pathways, including
β-catenin and MAPK, phosphatidylinositol-3 kinase/Akt and NF-κB
have been implicated in the induction of EMT in cancer cells. These
pathways are responsible for increased cell proliferation,
apoptosis, EMT, invasion, metastasis and chemoresistance in a
number of human cancers (28,29).
We evaluated the impact of Mcl-1 expression on oncogenic signaling
pathways. Our study showed that the β-catenin, MEK1/2, ERK1/2 and
p38 pathways were significantly blocked when Mcl-1 expression was
knocked down.
In summary, knockdown of Mcl-1 led to increased
adhesion of human GC cells to fibronectin and collagen I. Knockdown
of Mcl-1 inhibited EMT induction, as the expression levels of
vimentin, MMP-2, MMP-9 and Snail in human GC cells were decreased.
Additionally, knockdown of Mcl-1 suppressed tumor cell migration
and invasion. The β-catenin, MEK1/2, ERK1/2 and p38 pathways were
significantly blocked by knockdown of Mcl-1. These results revealed
that Mcl-1 expression induces EMT via the β-catenin, MEK1/2 and
MAPK signaling pathways, thereby stimulating the invasive and
migratory capacities of human GC cells.
Acknowledgments
This study was supported by research funds from the
Research Institute of Clinical Medicine, Chonnam National
University Hwasun Hospital, Republic of Korea in 2014 (HCRI
14028-21).
References
|
1
|
Kuwahara A, Takachi R, Tsubono Y, Sasazuki
S, Inoue M and Tsugane S; JPHC Study Group: Socioeconomic status
and gastric cancer survival in Japan. Gastric Cancer. 13:222–230.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Krejs GJ: Gastric cancer: Epidemiology and
risk factors. Dig Dis. 28:600–603. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tan IB, Ng I, Tai WM and Tan P:
Understanding the genetic basis of gastric cancer: recent advances.
Expert Rev Gastroenterol Hepatol. 6:335–341. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Llambi F and Green DR: Apoptosis and
oncogenesis: Give and take in the BCL-2 family. Curr Opin Genet
Dev. 21:12–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weyhenmeyer B, Murphy AC, Prehn JH and
Murphy BM: Targeting the anti-apoptotic Bcl-2 family members for
the treatment of cancer. Exp Oncol. 34:192–199. 2012.PubMed/NCBI
|
|
7
|
Davids MS and Letai A: Targeting the
B-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol.
30:3127–3135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thomas LW, Lam C and Edwards SW: Mcl-1;
the molecular regulation of protein function. FEBS Lett.
584:2981–2989. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Akgul C: Mcl-1 is a potential therapeutic
target in multiple types of cancer. Cell Mol Life Sci.
66:1326–1336. 2009. View Article : Google Scholar
|
|
10
|
Mandelin AM II and Pope RM: Myeloid cell
leukemia-1 as a therapeutic target. Expert Opin Ther Targets.
11:363–373. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Perciavalle RM and Opferman JT: Delving
deeper: MCL-1’s contributions to normal and cancer biology. Trends
Cell Biol. 23:22–29. 2013. View Article : Google Scholar
|
|
12
|
Zhang T, Zhao C, Luo L, Zhao H, Cheng J
and Xu F: The expression of Mcl-1 in human cervical cancer and its
clinical significance. Med Oncol. 29:1985–1991. 2012. View Article : Google Scholar
|
|
13
|
Luo L, Zhang T, Liu H, Lv T, Yuan D, Yao
Y, Lv Y and Song Y: miR-101 and Mcl-1 in non-small-cell lung
cancer: Expression profile and clinical significance. Med Oncol.
29:1681–1686. 2012. View Article : Google Scholar
|
|
14
|
Henderson-Jackson EB, Helm J, Ghayouri M,
Hakam A, Nasir A, Leon M, Bui M, Yeatman T and Coppola D:
Correlation between Mcl-1 and pAKT protein expression in colorectal
cancer. Int J Clin Exp Pathol. 3:768–774. 2010.PubMed/NCBI
|
|
15
|
Likui W, Qun L, Wanqing Z, Haifeng S,
Fangqiu L and Xiaojun L: Prognostic role of myeloid cell leukemia-1
protein (Mcl-1) expression in human gastric cancer. J Surg Oncol.
100:396–400. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maeta Y, Tsujitani S, Matsumoto S,
Yamaguchi K, Tatebe S, Kondo A, Ikeguchi M and Kaibara N:
Expression of Mcl-1 and p53 proteins predicts the survival of
patients with T3 gastric carcinoma. Gastric Cancer. 7:78–84. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsujitani S, Saito H, Wakatsuki T,
Ikeguchi M, Shirabe K, Morita M, Kakeji Y, Yano T and Maehara Y:
Relationship between expression of apoptosis-related proteins and
the efficacy of postoperative chemotherapy in patients with T3
gastric cancer. Surg Today. 42:225–232. 2012. View Article : Google Scholar
|
|
18
|
Steinestel K, Eder S, Schrader AJ and
Steinestel J: Clinical significance of epithelial-mesenchymal
transition. Clin Transl Med. 3:172014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Davis FM, Stewart TA, Thompson EW and
Monteith GR: Targeting EMT in cancer: Opportunities for
pharmacological intervention. Trends Pharmacol Sci. 35:479–488.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Peng Z, Wang CX, Fang EH, Wang GB and Tong
Q: Role of epithelial-mesenchymal transition in gastric cancer
initiation and progression. World J Gastroenterol. 20:5403–5410.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Natalwala A, Spychal R and Tselepis C:
Epithelial-mesenchymal transition mediated tumourigenesis in the
gastrointestinal tract. World J Gastroenterol. 14:3792–3797. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Townson JL and Chambers AF: Dormancy of
solitary metastatic cells. Cell Cycle. 5:1744–1750. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chambers AF, Groom AC and MacDonald IC:
Dissemination and growth of cancer cells in metastatic sites. Nat
Rev Cancer. 2:563–572. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ombrato L and Malanchi I: The EMT
universe: space between cancer cell dissemination and metastasis
initiation. Crit Rev Oncog. 19:349–361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Findlay VJ, Wang C, Watson DK and Camp ER:
Epithelial-to-mesenchymal transition and the cancer stem cell
phenotype: Insights from cancer biology with therapeutic
implications for colorectal cancer. Cancer Gene Ther. 21:181–187.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu X and Fan D: The
epithelial-mesenchymal transition and cancer stem cells: functional
and mechanistic links. Curr Pharm Des. 21:1279–1291. 2015.
View Article : Google Scholar
|
|
28
|
Lindsey S and Langhans SA: Crosstalk of
oncogenic signaling pathways during epithelial-mesenchymal
transition. Front Oncol. 4:3582014. View Article : Google Scholar
|
|
29
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|