Introduction
Esophageal squamous cell carcinoma (ESCC) is a
malignant disease with a poor prognosis. The standard treatment for
advanced ESCC patients has been surgical resection, although
patients with unresectable ESCC are usually treated by definitive
chemoradiotherapy (CRT) (1).
Various anticancer agents have been used to enhance the therapeutic
effects of radiotherapy (2–4), including cisplatin and 5-fluorouracil
(2,5,6).
Although favorable outcomes after CRT compared with radiation
therapy alone have been reported (1,7),
adverse events are also noted, such as pulmonary dysfunction,
leukocytopenia, radiation pneumonitis, pericardial effusion,
pleural effusion, perforation and stenosis (8,9). Thus,
there is a need to both enhance therapeutic effects and reduce
radiation-induced adverse events when performing CRT for esophageal
cancer.
Histone acetyltransferases (HATs) and histone
deacetylases (HDACs) regulate the acetylation and deacetylation of
histones, respectively. Core histone acetylation relaxes the
chromatin structure and facilitates transcription (10,11).
HDACs are involved in the deacetylation of chromatin histone
proteins as well as non-histone proteins, which regulate cell
differentiation, apoptosis and growth arrest (12,13).
HDAC inhibitors act additively or synergistically with conventional
cancer therapies such as radiotherapy, and numerous studies have
shown that they enhance the radiation sensitivity of various cancer
cell lines both in vitro and in vivo (14–23).
These effects have been explained as the modulation of cell cycle
regulation, particularly G1-phase arrest, the inhibition of DNA
synthesis, the suppression of DNA repair pathways, apoptosis and
the alteration of chromatin structures by histone hyperacetylation
(16,19,20,24).
Much attention has been given to the suppression of
the double-strand break (DSB) repair pathway by HDAC inhibitors,
which enhance the radiation sensitivity and accumulation of the
sensitive marker of DSBs, H2AX phosphorylation (γH2AX) (19). Homologous recombination (HR) and
non-homologous end joining (NHEJ) are the main pathways by which
radiation-induced DSBs are repaired (25–27).
Rad51 is a key protein of HR, while NHEJ involves the Ku70/Ku86
heterodimer which binds with strong affinity to broken DNA ends,
and recruits the catalytic subunit of DNA-dependent protein kinases
(DNA-PKcs). Ku70 functions are regulated by HATs and HDACs, and an
increase in Ku70 expression in cancer cells was found to enhance
the DNA DSB repair ability and reduce Bax-mediated apoptosis
(28). Ku70 is also a target of
some class I, II and III HDACs (29), while the use of HDAC inhibitors led
to increased Ku70 acetylation accompanied by a reduced DNA binding
affinity, with no disruption to Ku70/Ku80 heterodimer formation
(30). Treatment of a myelogenous
leukemia cell line with HDAC inhibitors before irradiation resulted
in increased and prolonged expression of γH2AX repair foci
(24).
Valproic acid (VPA), an 8-carbon branched-chain
fatty acid, has a well-established efficacy for the treatment of
epilepsy and other seizure disorders. VPA was also shown to be an
effective inhibitor of HDACs (31,32),
and to enhance radiosensitivity in various cancer cell lines
(18,33–35).
We recently reported a clinically safe dose of VPA that can enhance
radiation-induced cytotoxicity in human ESCC cells by chromatin
decondensation with histone hyperacetylation and the downregulation
of Rad51 (36). For certain HDAC
inhibitors, the acetylation of proteins in NHEJ has been
demonstrated to be involved in the inhibition of radiation-induced
DSBs repair (37). However, the
mechanism underlying the inhibitory effect of VPA with respect to
NHEJ in esophageal cancer has not yet been confirmed. The present
study therefore examined whether VPA inhibits radiation-induced DNA
DSB repair by suppressing NHEJ in human ESCC cells, focusing
particularly on the acetylation of Ku70 by VPA.
Materials and methods
Cell lines and cell culture
Three human ESCC cell lines were used in the present
study. The poorly differentiated ESCC cell line TE9, and the
moderately differentiated TE11 cells were provided by Dr Tetsuro
Nishihira (Higashimatsuyama Medical Association Hospital, Saitama,
Japan). The highly differentiated ESCC cell line KES was
established in our laboratory from endoscopic biopsy specimens
obtained from a patient carrying highly differentiated ESCC. These
cells were seeded in 75-cm2 dishes (Becton-Dickinson,
Tokyo, Japan) and cultured in 10 ml of medium consisted of
RPMI-1640 (Gibco®, Life Technologies Corp., Tokyo,
Japan) supplemented with 10% heat-inactivated fetal bovine serum
(Nichirei Biosciences, Inc., Tokyo, Japan), 100 IU/ml penicillin
and 100 µg/ml streptomycin (Life Technologies Corp.) at 37°C
in a humidified atmosphere of 5% CO2. Cells were grown
to confluency and harvested by trypsinization with 0.25 mg/ml
trypsin/EDTA (Life Technologies Corp.), then resuspended in culture
medium before use.
Reagents and antibodies
VPA was purchased from Sigma-Aldrich Co. (Tokyo,
Japan). Rabbit anti-phospho-histone H2AX (Ser 139) (γH2AX) (cat.
#9718) and anti-acetylatedlysine monoclonal antibodies (cat. #9814)
were obtained from Cell Signaling Technology Co. Ltd. (Tokyo,
Japan). Rabbit monoclonal anti-Ku70 antibody was purchased from
Abcam (cat. #ab92450; Cambridge, UK), and goat anti-rabbit IgG
horseradish peroxidase (HRP)-conjugated were purchased from Cell
Signaling Technology (cat. #7074). β-actin and TATA-binding protein
(TBP) were detected as control proteins using rabbit polyclonal
anti-β-actin antibody (cat. #4967) and rabbit polyclonal anti-TBP
antibody (cat. #8515) (both from Cell Signaling Technology),
respectively.
Irradiation
Cells were irradiated by MBR-1520R-3 (Hitachi
Medicotechnology, Hitachi, Japan) at a dose rate of 1 Gy/min. The X
ray-irradiation power output was 125 kV, 20 mA. Forward-scattered
radiation was filtered using a 0.5 mm Al and 0.2 mm Cu filter.
Immunofluorescent cytochemistry
Cells were cultured on Lab-Tec chamber slides (Nalge
Nunc International, Rochester, NY, USA), treated with or without
VPA (0.5 mM) for 24 h, then irradiated (6 Gy). Treated cells were
fixed in methanol and acetone (1:1) for 1 min after irradiation at
designated time-points (0/4/8 h). After washing in
phosphate-buffered saline (PBS), the slides were immersed in
methanol containing 0.3% H2O2 for 10 min,
blocked with 3.3% normal goat serum in PBS, and incubated with the
rabbit monoclonal anti-phospho-histone H2AX (Ser 139) antibody
(1:200) at 4°C overnight. Following three washes in PBS, slides
were incubated with an anti-rabbit IgG antibody conjugated with
Alexa Fluor® 594 (cat. #A11012; Molecular
Probes/Invitrogen, Carlsbad, CA, USA) for 1 h at room temperature
in the dark. Nuclei were stained with Hoechst 33258 for 5 min, and
slides were observed under an immunofluorescence microscope
(Olympus BX50/BX-FLA; Tokyo, Japan).
Quantification of γH2AX levels after
irradiation
To assess changes in radiation-induced DNA DSB
repair, we compared γH2AX levels in irradiated cells treated with
or without VPA (0.5 mM) using western blotting. Cells treated with
combination therapy were pre-treated with VPA for 24 h before
irradiation. Cell lysates were collected after irradiation at
designated times (0/10/30 min/1/2/4/8 h). The protein concentration
in each lysate was measured using the Pierce BCA protein assay kit
(Thermo Fisher Scientific, Rockford, IL, USA). A sample volume
equivalent to 20 µg protein from each lysate was loaded on a
10% polyacrylamide gel (Mini-Protean TGX Precast Gels; Bio-Rad
Laboratories Inc., Berkeley, CA, USA) and separated by
electrophoresis. Proteins were transferred to polyvinylidene
difluoride (PVDF) membranes (Trans-Blot Turbo mini-PVDF Transfer
Pack; Bio-Rad Laboratories Inc.), blocked with Tris-buffered saline
with Tween-20 Tablets, pH 7.6 (Takara Bio, Inc., Shiga, Japan),
then incubated with the rabbit monoclonal anti-phospho-histone H2AX
(Ser 139) primary antibody at a 1:1,000 dilution for 10 min at room
temperature. A rabbit polyclonal β-actin antibody at 1:5,000
dilution was used to detect β-actin as a control protein. Secondary
antibody incubation using goat anti-rabbit IgG HRP-conjugated at a
1:4,000 dilution was carried out according to the manufacturer's
instructions for 10 min at room temperature (cat. SNAP2BASE; SNAP
i.d. 2.0; Millipore, Billerica, MA, USA). Membranes were treated
with PVDF Blocking Reagent for Can Get Signal® (Toyobo
Co., Ltd., Osaka, Japan). The antibody-antigen complex was detected
using enhanced chemiluminescence by an ECL western blotting
detection kit (General Electric Healthcare Japan Co., Ltd., Tokyo,
Japan) and the Light-Capture system (ATTO Co., Ltd., Tokyo, Japan).
Quantification was performed by the CS analyzer program (ATTO Co.,
Ltd.) and normalized to β-actin levels.
Assessment of Ku70 acetylation
Cells were incubated with or without VPA (0.5 mM)
for 24 h, then irradiated (6 Gy) and harvested promptly. Nuclear
proteins were extracted using a Nuclear Extract kit (Active Motif,
Tokyo, Japan), and protein concentrations were determined using a
Pierce BCA protein assay kit as before. A rabbit monoclonal
anti-Ku70 antibody at 1:80 dilution was used for
immunoprecipitation. Magnetic beads (Dynabeads® Protein
G Immunoprecipitation kit; Life Technologies, Carlsbad, CA, USA)
were used for immunoprecipitation of the target antigen according
to the manufacturer's instructions. The target antibody was
incubated with Dynabeads® with rotation for 10 min to
enable binding to occur. After adding the extracted nuclear protein
to the beads-antibody complex, the sample was incubated at 4°C
overnight with rotation to form an antigen-beads-antibody complex.
The target antigen was dissociated from the beads-antibody complex
with elution buffer. Ku70 acetylation was then examined by western
blotting using a rabbit monoclonal anti-acetylated-lysine primary
antibody at a 1:300 dilution. To detect TBP as a control nuclear
protein, a rabbit polyclonal anti-TBP antibody (Cell Signaling
Technology) at 1:300 dilution was used. Goat anti-rabbit IgG-HRP
conjugates at 1:4,000 dilution were used as secondary
antibodies.
Results
Changes of radiation-induced γH2AX levels
by VPA
γH2AX levels in cells pre-treated with VPA were
higher than in untreated cells in all three ESCC cell lines
(Fig. 1). γH2AX levels were
detected by western blotting and quantified by measuring the ratio
of γH2AX to β-actin. The time course of γH2AX is displayed in
Fig. 2. VPA was shown to prolong
γH2AX levels after irradiation in all three ESCC cell lines.
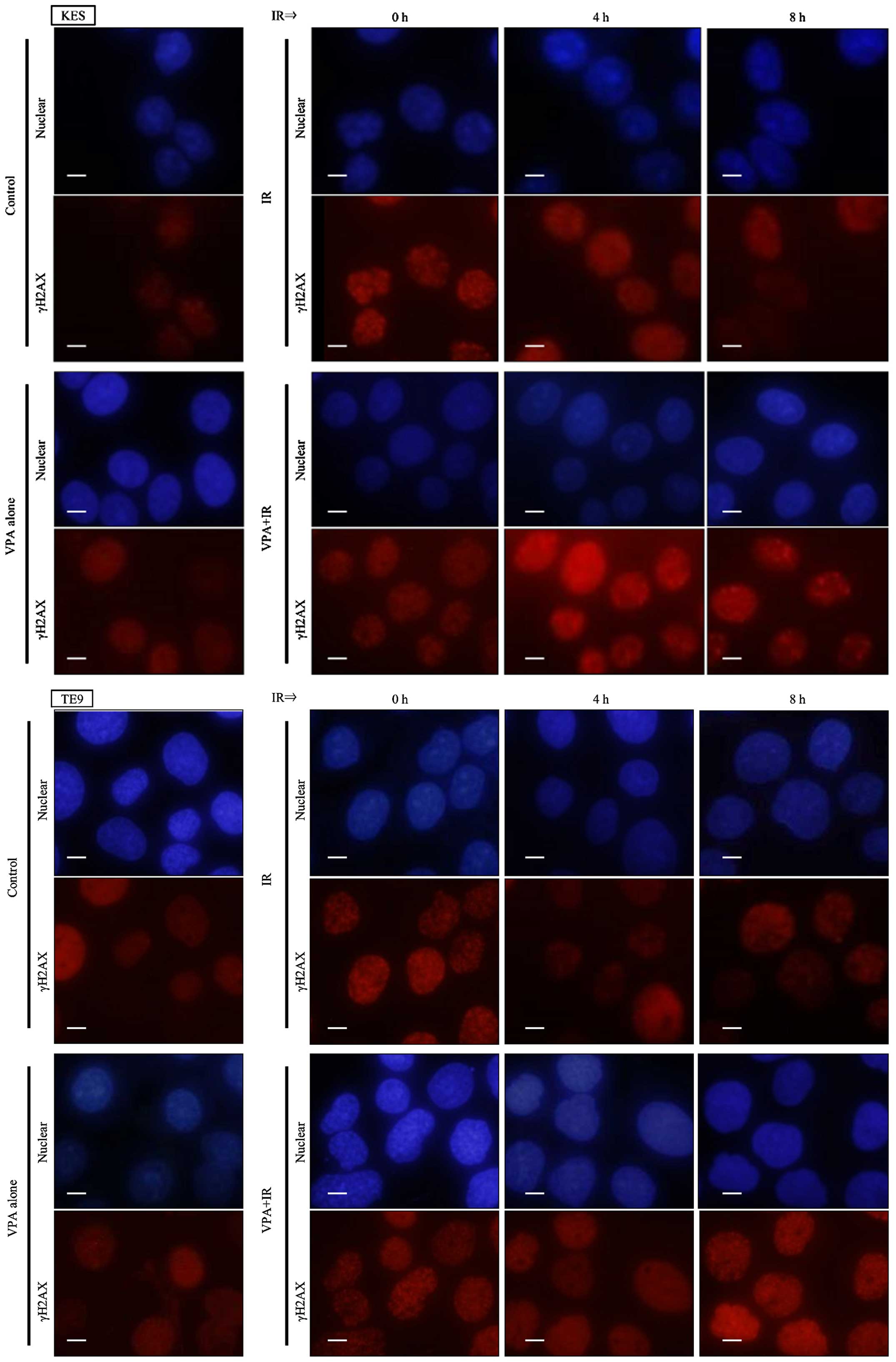
Moreover, prolonged γH2AX foci formation after irradiation was also
observed by immunocytochemistry following VPA pretreatment in KES
and TE9 cells (Fig. 3).
Acetylation of Ku70 by VPA
The acetylation of Ku70 by VPA was estimated by
western blotting following immunoprecipitation of the nuclear
extract with an anti-Ku70 antibody and the detection of
acetylated-lysine. In all three ESCC cell lines, increased Ku70
acetylation after irradiation was observed following pretreatment
with 0.5 mM VPA for 24 h (Fig.
4).
Discussion
In the present study, we showed that VPA inhibited
the radiation-induced DNA DSB repair in ESCC cell lines, and that
this was accompanied by acetylation of the DSB repair protein,
Ku70.
Previously, γH2AX has been described as a highly
sensitive method to detect irradiation-induced DSBs (38,39).
In response to this, serine 139 within the conserved C-terminal
region of the H2AX protein becomes phosphorylated by one of the DNA
damage signaling kinases of the
phosphatidylinositol-3-OH-kinase-like family. This phosphorylation
rapidly spreads to a region of chromatin surrounding the DSBs,
resulting in the formation of γH2AX foci. The main role of H2AX
phosphorylation in the DNA damage response was reported to be the
recruitment of DNA repair proteins to DSB sites and the preparation
of a platform for signaling and repair (38,40).
In the present study, we analyzed H2AX phosphorylation levels by
western blotting, and examined γH2AX foci by immunocytochemistry as
a marker of DNA DSB after irradiation.
More than 18 mammalian HDAC enzymes (HDAC 1–11 and
sirtuins 1–7) have been identified, which are grouped into four
classes according to their homology (41). HDAC inhibitors are an emerging class
of drugs that have shown promise as anticancer agents when used
alone or in combination with conventional therapies (42,43).
VPA has been used for the treatment of seizure disorders and
epilepsy. Previous studies found that it induces apoptosis and the
accumulation of hyperacetylated histones H3 and H4, and also
inhibits class I HDACs (HDACs 1, 2 and 3) and class II subclass I
HDACs (HDACs 4, 5 and 7) (31,32,44,45).
Therapeutic plasma concentrations used clinically for epilepsy
treatment range from 50–100 µg/ml, which is equivalent to
0.3–0.6 mM. Therefore, 0.5 mM VPA is a clinically safe
concentration (46) that we used in
the present study based on our previous growth inhibition assay in
which 0.5 mM VPA did not inhibit the growth of ESCC cells (36).
We recently showed that VPA enhanced the
radiosensitizing effect by chromatin decondensation with histone
hyperacetylation and downregulation of Rad51 (36). Various mechanisms are responsible
for this VPA-mediated enhancement in radiosensitivity. Firstly,
HDAC inhibitors lead to the acetylation of histone tails, which
induces reduction of the electrostatic charge interactions between
DNA and histones, permitting access to the chromatin structure in
association with chromatin decondensation (47,48).
Additionally, Harikrishnan et al (49) reported that euchromatin was more
sensitive to radiation-induced DSBs than heterochromatin, and that
histone modifications contributed to the radiosensitivity effects
of HDAC inhibitors. Thus, they concluded that heterochromatin is
more resistant to histone modification and DNA damage. Secondly,
HDAC inhibitors inhibit the radiation-induced DNA DSB repair
process. In the present study, we confirmed that VPA prolonged
radiation-induced DNA DSB repair by measuring γH2AX level changes
overtime after irradiation. This lengthening of DNA DSBs can be
attributed to the VPA-induced inhibition of the DSB repair process.
While we previously found that Rad51 expression was downregulated
by VPA, and Adimoolam et al (37) reported that suberoylanilide
hydroxamic acid (PCI-24781) also suppressed Rad51 expression, the
mechanism of its suppression by HDAC inhibitors has not been fully
clarified. Further studies are therefore necessary to understand
this.
The functional inhibition of non-histone proteins by
HDAC inhibitors has recently been reported to involve acetylation
(30). However, it is difficult to
examine the acetylation of non-histone proteins by western blotting
due to the lack of an appropriate primary antibody. Recent studies
reported that the acetylation of non-histone proteins by HDAC
inhibitors induced the functional inhibition of DNA repair activity
(20,30). Furthermore, Chen et al
(30) found that HDAC inhibitors
reduced the DNA end-binding affinity of Ku70 by acetylation. In the
present study, we focused on Ku70 as a key protein of NHEJ in the
radiation-induced DNA DSB repair pathway. We confirmed that VPA
increased Ku70 acetylation levels in three ESCC cell lines.
In DNA DSB repair pathways, ataxia telangiectasia
mutated and ataxia telangiectasia related proteins are important
components of the DSB signaling cascade. Their activation results
in the phosphorylation of downstream substrates, including p53
(50). HDACs can regulate the
expression of various genes by direct interaction (13). For instance, p53 is downregulated by
HDACs 1, 2 and 3 (35,51) and acetylated by VPA (45), which induces cell cycle regulation,
apoptosis and inhibition of DNA DSB repair (45,50).
Several proteins inhibit DNA DSBs repair pathways by mediating the
resection process of DNA ends (52,53).
Thus, VPA inhibits HR in the repair pathway by acetylating Sae2 and
Exo1, which are two nucleases involved in resecting DNA ends
(54). Our present findings and
those of earlier studies showed that VPA has the potential to
synergistically inhibit DNA DSB repair pathways by acetylating p53
in the DSB signaling cascade, nucleases involved in DNA end
resection and Ku70 in NHEJ, as well as suppressing Rad51 expression
levels in HR. VPA may therefore be a suitable agent to combine with
radiotherapy due to its multiple effects in DNA DSB repair
inhibition. Further studies are needed to clarify the precise
mechanisms of DNA DSB repair inhibition by VPA in ESCC.
Tomita et al observed a difference in the
proportion of HR and NHEJ in DSB repair in chicken B lymphoid cell
lines according to the irradiation dose rate. Although both HR and
NHEJ were activated following high-dose irradiation (0.9 Gy/min),
the activation of NHEJ dominated after low-dose irradiation (1.0
mGy/min) (55). This suggests that
the inhibition of NHEJ may be a more effective means of enhancing
radiosensitivity, regardless of the radiation dose. Moreover,
Karagiannis and El-Osta (50)
reported a radiation protection effect associated with HDAC
inhibitors. These not only act as radiosensitizers in response to
irradiation, but also reduce radiation-induced dermatitis and
esophagitis by suppressing the expression of transforming growth
factor-β and tumor necrosis factor-α. Therefore, the concomitant
use of VPA with radiation therapy against esophageal cancer has the
potential to reduce various radiation-induced adverse effects.
In conclusion, VPA treatment at clinically safe
concentrations enhances radiosensitivity by inhibiting both HR and
NHEJ in the DNA DSB repair process, and prolonging DNA DSBs.
Therefore, VPA has considerable potential as a therapeutic agent
for esophageal cancer.
Acknowledgments
We are grateful to Medical Technologist of
Gastroenterologic Surgery, Kanazawa University for their important
contributions to the experiments.
Abbreviations:
|
DSBs
|
DNA double-strand breaks
|
|
ESCC
|
esophageal squamous cell carcinoma
|
|
γH2AX
|
H2AX phosphorylation
|
|
HDACs
|
histone deacetylases
|
|
HR
|
homologous recombination
|
|
NHEJ
|
non-homologous end joining
|
|
VPA
|
valproic acid
|
References
|
1
|
Herskovic A, Martz K, al-Sarraf M,
Leichman L, Brindle J, Vaitkevicius V, Cooper J, Byhardt R, Davis L
and Emami B: Combined chemotherapy and radiotherapy compared with
radiotherapy alone in patients with cancer of the esophagus. N Engl
J Med. 326:1593–1598. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohtsu A, Boku N, Muro K, Chin K, Muto M,
Yoshida S, Satake M, Ishikura S, Ogino T, Miyata Y, et al:
Definitive chemoradiotherapy for T4 and/or M1 lymph node squamous
cell carcinoma of the esophagus. J Clin Oncol. 17:2915–2921.
1999.PubMed/NCBI
|
|
3
|
Makino I, Ninomiya I, Okamoto K, Kinoshita
J, Hayashi H, Nakamura K, Oyama K, Nakagawara H, Fujita H, Tajima
H, et al: A pilot study of chemoradiotherapy with weekly docetaxel
for thoracic esophageal carcinoma with T4 and/or M1 lymph node
metastasis. World J Oncol. 2:252–258. 2011.
|
|
4
|
van Hagen P, Hulshof MC, van Lanschot JJ,
Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BP, Richel DJ,
Nieuwenhuijzen GA, Hospers GA, Bonenkamp JJ, et al CROSS Group:
Preoperative chemoradiotherapy for esophageal or junctional cancer.
N Engl J Med. 366:2074–2084. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaneko K, Ito H, Konishi K, Kurahashi T,
Ito T, Katagiri A, Yamamoto T, Kitahara T, Mizutani Y, Ohtsu A, et
al: Definitive chemoradiotherapy for patients with malignant
stricture due to T3 or T4 squamous cell carcinoma of the
oesophagus. Br J Cancer. 88:18–24. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ishida K, Ando N, Yamamoto S, Ide H and
Shinoda M: Phase II study of cisplatin and 5-fluorouracil with
concurrent radiotherapy in advanced squamous cell carcinoma of the
esophagus: A Japan Esophageal Oncology Group (JEOG)/Japan Clinical
Oncology Group trial (JCOG9516). Jpn J Clin Oncol. 34:615–619.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cooper JSG, Guo MD, Herskovic A, Macdonald
JS, Martenson JA Jr, Al-Sarraf M, Byhardt R, Russell AH, Beitler
JJ, Spencer S, et al Radiation Therapy Oncology Group:
Chemoradiotherapy of locally advanced esophageal cancer: Long-term
follow-up of a prospective randomized trial (RTOG 85-01). JAMA.
281:1623–1627. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ishikura S, Nihei K, Ohtsu A, Boku N,
Hironaka S, Mera K, Muto M, Ogino T and Yoshida S: Long-term
toxicity after definitive chemoradiotherapy for squamous cell
carcinoma of the thoracic esophagus. J Clin Oncol. 21:2697–2702.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kato K, Muro K, Minashi K, Ohtsu A,
Ishikura S, Boku N, Takiuchi H, Komatsu Y, Miyata Y and Fukuda H;
Gastrointestinal Oncology Study Group of the Japan Clinical
Oncology Group (JCOG): Phase II study of chemoradiotherapy with
5-fluorouracil and cisplatin for Stage II–III esophageal squamous
cell carcinoma: JCOG trial (JCOG 9906). Int J Radiat Oncol Biol
Phys. 81:684–690. 2011. View Article : Google Scholar
|
|
10
|
Vettese-Dadey M, Grant PA, Hebbes TR,
Crane-Robinson C, Allis CD and Workman JL: Acetylation of histone
H4 plays a primary role in enhancing transcription factor binding
to nucleosomal DNA in vitro. EMBO J. 15:2508–2518. 1996.PubMed/NCBI
|
|
11
|
Ura K, Kurumizaka H, Dimitrov S, Almouzni
G and Wolffe AP: Histone acetylation: Influence on transcription,
nucleosome mobility and positioning, and linker histone-dependent
transcriptional repression. EMBO J. 16:2096–2107. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lindemann RK, Gabrielli B and Johnstone
RW: Histonedeacetylase inhibitors for the treatment of cancer. Cell
Cycle. 3:779–788. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ropero S and Esteller M: The role of
histone deacetylases (HDACs) in human cancer. Mol Oncol. 1:19–25.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Camphausen K, Burgan W, Cerra M, Oswald
KA, Trepel JB, Lee MJ and Tofilon PJ: Enhanced radiation-induced
cell killing and prolongation of gammaH2AX foci expression by the
histone deacetylase inhibitor MS-275. Cancer Res. 64:316–321. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Camphausen K, Scott T, Sproull M and
Tofilon PJ: Enhancement of xenograft tumor radiosensitivity by the
histone deacetylase inhibitor MS-275 and correlation with histone
hyperacetylation. Clin Cancer Res. 10:6066–6071. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y and Jung M, Dritschilo A and Jung
M: Enhancement of radiation sensitivity of human squamous carcinoma
cells by histone deacetylase inhibitors. Radiat Res. 161:667–674.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chinnaiyan P, Vallabhaneni G, Armstrong E,
Huang SM and Harari PM: Modulation of radiation response by histone
deacetylase inhibition. Int J Radiat Oncol Biol Phys. 62:223–229.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Camphausen K, Cerna D, Scott T, Sproull M,
Burgan WE, Cerra MA, Fine H and Tofilon PJ: Enhancement of in vitro
and in vivo tumor cell radiosensitivity by valproic acid. Int J
Cancer. 114:380–386. 2005. View Article : Google Scholar
|
|
19
|
Karagiannis TC, Harikrishnan KN and
El-Osta A: The histone deacetylase inhibitor, trichostatin A,
enhances radiation sensitivity and accumulation of gammaH2A.X.
Cancer Biol Ther. 4:787–793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Munshi A, Kurland JF, Nishikawa T, Tanaka
T, Hobbs ML, Tucker SL, Ismail S, Stevens C and Meyn RE: Histone
deacetylase inhibitors radiosensitize human melanoma cells by
suppressing DNA repair activity. Clin Cancer Res. 11:4912–4922.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jung M, Velena A, Chen B, Petukhov PA,
Kozikowski AP and Dritschilo A: Novel HDAC inhibitors with
radiosensitizing properties. Radiat Res. 163:488–493. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen X, Wong P, Radany E and Wong JY: HDAC
inhibitor, valproic acid, induces p53-dependent radiosensitization
of colon cancer cells. Cancer Biother Radiopharm. 24:689–699. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang B, Wang Y and Pang X: Enhanced
radiosensitivity of EC109 cells by inhibition of HDAC1 expression.
Med Oncol. 29:340–348. 2012. View Article : Google Scholar
|
|
24
|
Karagiannis TC, Harikrishnan KN and
El-Osta A: Disparity of histone deacetylase inhibition on repair of
radiation-induced DNA damage on euchromatin and constitutive
heterochromatin compartments. Oncogene. 26:3963–3971. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mahaney BL, Meek K and Lees-Miller SP:
Repair of ionizing radiation-induced DNA double-strand breaks by
non-homologous end-joining. Biochem J. 417:639–650. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dobbs TA, Tainer JA and Lees-Miller SP: A
structural model for regulation of NHEJ by DNA-PKcs
autophosphorylation. DNA Repair. 9:1307–1314. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Botrugno OA, Robert T, Vanoli F, Foiani M
and Minucci S: Molecular pathways: Old drugs define new pathways:
Non-histone acetylation at the crossroads of the DNA damage
response and autophagy. Clin Cancer Res. 18:2436–2442. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cohen HY, Lavu S, Bitterman KJ, Hekking B,
Imahiyerobo TA, Miller C, Frye R, Ploegh H, Kessler BM and Sinclair
DA: Acetylation of the C terminus of Ku70 by CBP and PCAF controls
Bax-mediated apoptosis. Mol Cell. 13:627–638. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hassan MK, Watari H, Salah-Eldin AE,
Sultan AS, Mohamed Z, Fujioka Y, Ohba Y and Sakuragi N: Histone
deacetylase inhibitors sensitize lung cancer cells to hyperthermia:
Involvement of Ku70/SirT-1 in thermo-protection. PLoS One.
9:e942132014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen CS, Wang YC, Yang HC, Huang PH, Kulp
SK, Yang CC, Lu YS, Matsuyama S, Chen CY and Chen CS: Histone
deacetylase inhibitors sensitize prostate cancer cells to agents
that produce DNA double-strand breaks by targeting Ku70
acetylation. Cancer Res. 67:5318–5327. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Phiel CJ, Zhang F, Huang EY, Guenther MG,
Lazar MA and Klein PS: Histone deacetylase is a direct target of
valproic acid, a potent anticonvulsant, mood stabilizer, and
teratogen. J Biol Chem. 276:36734–36741. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Göttlicher M, Minucci S, Zhu P, Krämer OH,
Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG,
et al: Valproic acid defines a novel class of HDAC inhibitors
inducing differentiation of transformed cells. EMBO J.
20:6969–6978. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karagiannis TC, Kn H and El-Osta A: The
epigenetic modifier, valproic acid, enhances radiation sensitivity.
Epigenetics. 1:131–137. 2006. View Article : Google Scholar
|
|
34
|
Chinnaiyan P, Cerna D, Burgan WE, Beam K,
Williams ES, Camphausen K and Tofilon PJ: Postradiation
sensitization of the histone deacetylase inhibitor valproic acid.
Clin Cancer Res. 14:5410–5415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen X, Wong JY, Wong P and Radany EH:
Low-dose valproic acid enhances radiosensitivity of prostate cancer
through acetylated p53-dependent modulation of mitochondrial
membrane potential and apoptosis. Mol Cancer Res. 9:448–461. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shoji M, Ninomiya I, Makino I, Kinoshita
J, Nakamura K, Oyama K, Nakagawara H, Fujita H, Tajima H, Takamura
H, et al: Valproic acid, a histone deacetylase inhibitor, enhances
radiosensitivity in esophageal squamous cell carcinoma. Int J
Oncol. 40:2140–2146. 2012.PubMed/NCBI
|
|
37
|
Adimoolam S, Sirisawad M, Chen J, Thiemann
P, Ford JM and Buggy JJ: HDAC inhibitor PCI-24781 decreases RAD51
expression and inhibits homologous recombination. Proc Natl Acad
Sci USA. 104:19482–19487. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vandersickel V, Depuydt J, Van Bockstaele
B, Perletti G, Philippe J, Thierens H and Vral A: Early increase of
radiation-induced γH2AX foci in a human Ku70/80 knockdown cell line
characterized by an enhanced radiosensitivity. J Radiat Res.
51:633–641. 2010. View Article : Google Scholar
|
|
39
|
Mah LJ, Orlowski C, Ververis K, Vasireddy
RS, El-Osta A and Karagiannis TC: Evaluation of the efficacy of
radiation-modifying compounds using γH2AX as a molecular marker of
DNA double-strand breaks. Genome Integr. 2:32011. View Article : Google Scholar
|
|
40
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Barneda-Zahonero B and Parra M: Histone
deacetylases and cancer. Mol Oncol. 6:579–589. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Erlich RB, Rickwood D, Coman WB, Saunders
NA and Guminski A: Valproic acid as a therapeutic agent for head
and neck squamous cell carcinomas. Cancer Chemother Pharmacol.
63:381–389. 2009. View Article : Google Scholar
|
|
43
|
Tan J, Cang S, Ma Y, Petrillo RL and Liu
D: Novel histone deacetylase inhibitors in clinical trials as
anti-cancer agents. J Hematol Oncol. 3:52010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gurvich N, Tsygankova OM, Meinkoth JL and
Klein PS: Histone deacetylase is a target of valproic acid-mediated
cellular differentiation. Cancer Res. 64:1079–1086. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Duenas-Gonzalez A, Candelaria M,
Perez-Plascencia C, Perez-Cardenas E, de la Cruz-Hernandez E and
Herrera LA: Valproic acid as epigenetic cancer drug: Preclinical,
clinical and transcriptional effects on solid tumors. Cancer Treat
Rev. 34:206–222. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wolff JE, Kramm C, Kortmann RD, Pietsch T,
Rutkowski S, Jorch N, Gnekow A and Driever PH: Valproic acid was
well tolerated in heavily pretreated pediatric patients with
high-grade glioma. J Neurooncol. 90:309–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Garcia-Ramirez M, Rocchini C and Ausio J:
Modulation of chromatin folding by histone acetylation. J Biol
Chem. 270:17923–17928. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim MS, Blake M, Baek JH, Kohlhagen G,
Pommier Y and Carrier F: Inhibition of histone deacetylase
increases cytotoxicity to anticancer drugs targeting DNA. Cancer
Res. 63:7291–7300. 2003.PubMed/NCBI
|
|
49
|
Harikrishnan KN, Karagiannis TC, Chow MZ
and El-Osta A: Effect of valproic acid on radiation-induced DNA
damage in euchromatic and heterochromatic compartments. Cell Cycle.
7:468–476. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Karagiannis TC and El-Osta A: The paradox
of histone deacetylase inhibitor-mediated modulation of cellular
responses to radiation. Cell Cycle. 5:288–295. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Juan LJ, Shia WJ, Chen MH, Yang WM, Seto
E, Lin YS and Wu CW: Histone deacetylases specifically
down-regulate p53-dependent gene activation. J Biol Chem.
275:20436–20443. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mimitou EP and Symington LS: Sae2, Exo1
and Sgs1 collaborate in DNA double-strand break processing. Nature.
455:770–774. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kaidi A, Weinert BT, Choudhary C and
Jackson SP: Human SIRT6 promotes DNA end resection through CtIP
deacetylation. Science. 329:1348–1353. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shubassi G, Robert T, Vanoli F, Minucci S
and Foiani M: Acetylation: A novel link between double-strand break
repair and autophagy. Cancer Res. 72:1332–1335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tomita M, Morohoshi F, Matsumoto Y, Otsuka
K and Sakai K: Role of DNA double-strand break repair genes in cell
proliferation under low dose-rate irradiation conditions. J Radiat
Res. 49:557–564. 2008. View Article : Google Scholar : PubMed/NCBI
|