Introduction
Epidermal growth factor receptor (EGFR) signaling is
crucial for cell survival, growth, proliferation and migration
(1,2). Dysregulated EGFR signaling correlates
with proliferation, invasiveness and drug resistance of multiple
types of cancer, such as brain, lung, and breast cancer (3,4). EGFR
activation triggers cascades of multiple downstream signaling,
including the phosphoinositide 3-kinase/AKT (PI3K/AKT) pathway,
mitogen-activated protein kinase (MAPK) pathway and signal
transducer and activator of transcription (STAT) pathway (2).
Among these pathways activated by EGFR, AKT
signaling regulates many cellular functions including growth,
survival, and invasiveness, its excessive activation plays
important roles in cancer. EGFR-AKT signaling was involved in
esophageal squamous cell carcinoma (ESCC) cell proliferation and
migration (5). EGF-induced AKT
signaling was also reported in ovarian cancer cell proliferation
(6). PI3K/AKT signaling cascades
may be responsible for EGF-activated MMP7 and consequent cancer
invasiveness in human gastric carcinoma cell lines SNU-5 and HGC27
(7). In non-small cell lung cancer
(NSCLC) cells EGF activated MMP9 via AKT signaling to promote NSCLC
invasiveness (8). Activation of
EGFR/PI3K/AKT pathway can also mediate hypoxia-induced drug
resistance in hepatocellular carcinoma cells (9).
Inhibition of the EGF-induced EGFR/AKT activation
was extensively used to promote cell apoptosis, suppress tumor cell
growth and invasiveness. For example, in OVCAR-3 ovarian cancer
cells, using PI3K inhibitors, but not ERK inhibitor, decreased
EGF-induced cellular viability, revealing the important role of AKT
pathway in EGF-induced cellular viability (6). AKT inhibitor significantly inhibited
the EGF-induced activation of MMP7 in gastric cancer cells
(7). PI3K inhibitor wortmannin
inhibited the EGF-induced EGFR/PI3K/AKT signaling pathway
concomitantly with inhibition of cell proliferation and cell cycle
progression of primary external auditory canal keratinocytes
(EACKs) (10).
Due to the important role of EGFR/AKT pathway
activation in tumor cell malignant phenotype, regulation on
EGFR/AKT pathway in vivo was studied by several groups
(11–13). Phosphatases were reported as potent
regulators of EGF-induced AKT signaling (13). Phosphatase PTPN2 (T-cell protein
tyrosine phosphatase; also known as TC-PTP) inhibited EGF-induced
AKT phosphorylation, but not ERK1/2 phosphorylation (12). Phosphatase PTPRJ (protein tyrosine
phosphatase receptor type J; also known as DEP-1/CD148) was found
to regulate EGF-induced PI3K and AKT activation (13). Among phosphatases, PTEN (phosphatase
and tensin homologue deleted on chromosome 10), the most effective
phosphatase, can also regulate EGF-induced AKT activation (13). However, how PTEN regulated
EGF-induced AKT activation was not completely elucidated.
PTEN was found to inhibit the activation of the
growth factor receptor PDGFR pathway, especially the activation of
PDGF-induced AKT pathway (14). One
of its mechanisms was via ternary complex PTEN/EBP50/PDGFR mediated
by EBP50. EBP50 (Ezrin-radixin-moesin-binding phosphoprotein-50,
also called NHERF) was reported to bind with PDGFR (15). EBP50 can also interact with PTEN
(14,16). So EBP50 could act as a bridge,
recruit PTEN to PDGFR at plasma membrane (PM) and restrict the
activation of the PI3K/AKT signaling. EBP50 expression also confers
susceptibility to PDGFR pharmacological inhibition in breast cancer
(14,17). EBP50 mutations disrupting the
PTEN/EBP50/PDGFR complex formation would inactivate the inhibition
of EBP50 on PDGF-induced AKT activation. Both EBP50 K172N and D301V
mutations found in breast cancer can abolish PTEN/EBP50/PDGFR
complex formation, and retard the inhibition of PTEN on
PDGF-induced AKT activation (18),
revealing an emerging model regulating growth factor-mediated AKT
signaling-adaptor protein bound with growth factor receptor and
further mediated PTEN binding with growth factor receptor (14,19).
Both our previous research results, and those of
other groups revealed that EGFR can bind to EBP50 (20,21),
and PTEN also interacted with EBP50 (14,16),
providing the rationale that EBP50 can organize the complex of PTEN
and EGFR to regulate the EGFR/AKT pathway. Thus, in this study we
first examined the formation of PTEN, EBP50 and EGFR complex by
using co-immunoprecipitation (Co-IP) and GST pull-down assays.
Further, we investigated the effect of their association on
EGFR/AKT pathway by abolishing the complex formation. Results
showed that EBP50 specifically scaffolds the interaction of PTEN
with EGFR and enhances the inhibition of PTEN on EGF-induced AKT
activation. These results elucidated a novel mechanism regulating
EGF-induced AKT signaling.
Materials and methods
Preparation of plasmids
Glutathione S-transferase (GST)-tagged PTEN
carboxyl-terminal (GST-PTEN-CT, encoding amino acid from 374 to
403) plasmid was generated via polymerase chain reaction (PCR)
amplification of human PTEN cDNA, then inserted into pGEX-4T-1. The
inserts were verified by DNA sequencing. Isopropyl
β-D-1-Thiogalactopyranoside (IPTG) was used to induce the
expression of fusion proteins.
pBK-CMV-hemagglutinin (HA)-EBP50 expression plasmid
was kindly provided by Dr Randy Hall from Emory University
(Atlanta, GA, USA). pSuper.puro EBP50 shRNA and pSuper.puro
luciferase control shRNA plasmids were kindly provided by Dr
Margaret J. Wheelock from University of Nebraska Medical Center
(omaha, NE, USA). Full-length PTEN was inserted into pBK-CMV-Flag
vector for Flag-tagging. pBK-CMV-Flag-EGFR was kindly provided by
Dr Howard A. Rockman from Duke University Medical Center (Durham,
NC, USA).
PTEN carboxyl-terminal (CT) PDZ protein binding
motif point mutation (V403A-the last amino acid V403 was mutated to
A) and EGFR PDZ binding motif point mutation (L1043/1063F-key
residue L1043 and L1063 of PDZ binding site were mutated to F) were
created by PCR amplification and confirmed by sequencing.
Cell culture, transfection and cell
treatments
African green monkey kidney cell line COS-7 and
human astrocytoma U-373 MG (U373) cell lines (American Type Culture
Collection, ATCC; Manassas, VA, USA) were grown in Dulbecco's
modified Eagle's medium (DMEM; Gibco) and RPMI-1640 medium,
respectively. Both media contained 10% fetal bovine serum (FBS;
Hyclone, Logan, UT, USA) and 1% antibiotic-antimycotic agent (Life
Technologies, Inc., Grand Island, NY, USA). Cells were grown at
37°C and 5% CO2 to 80% confluency for use.
Transfections were performed by Lipofectamine 2000
(Invitrogen, Carlsbad, CA, USA) with plasmids DNA following the
protocol as reported before (22).
COS-7 or U373 cells were serum starved overnight,
then treated with 100 ng/ml EGF (Sigma-Aldrich, St. Louis, MO, USA)
for different time periods at 37°C (20) to detect the effect of PDZ binding
motif-mutated PTEN or EGFR overexpression on EGFR-mediated signal
transduction pathway. The effect of EBP50 knockdown on
EGFR-mediated signal transduction pathway was performed in the same
way.
Stable transfection
For EBP50 stable knockdown, COS-7 cells were
transfected with pSuper.puro EBP50 shRNA or control pSuper.puro
luciferase shRNA plasmid, respectively using Hifectin II (Applygen
Technologies Inc., Beijing, China) following the protocol. Two days
following transfection, cells were transferred to 90-mm plates and
cultured in selection medium with 0.5 µg/ml puromycin
(Sigma-Aldrich) to knock down EBP50 (20). The medium was changed every two days
to remove floating dead cells, and the resistant colonies formed
were harvested and plated in 24-well plates. Cell cultures were
expanded and cultured for at least a month, then the fractions were
used for analysis of EBP50 expression by western blotting, with
GAPDH expression as a protein loading control. Stably-transfected
cells were maintained and passaged in culture medium with puromycin
(0.25 µg/ml) (20).
siRNA-mediated transient EBP50
knockdown
Small interfering RNA (siRNA) duplexes directed
against EBP50 (nucleotides to: 5′-GUCGACCACCAGCAGGCGCACGGCGUUG-3′)
and control scrambled RNAi (5′-UCCAGACGGCGCAGUGGGCGACCGCUAC-3′)
were synthesized by Sigma-Aldrich. COS-7 cells were grown to 80%
confluency in 35-mm dishes, transfected with 2 µl
Lipofectamine 2000 (Invitrogen), and mixed with 36 pmol of the
synthetic EBP50 siRNA. The cells were then serum starved overnight,
stimulated, harvested and analyzed after 48 h of transfection.
Western blotting
Samples were run on 8% sodium dodecyl sulfate
(SDS)-polyacrylamide gels (PAGE) and transferred to PVDF membranes.
The blots were blocked in blocking buffer (5% non-fat dry milk in
TBST buffer) for 1 h at room temperature and then incubated with
primary antibodies in blocking buffer overnight at 4°C. The blots
were washed three times with TBST buffer and incubated for 1 h at
room temperature with a horseradish peroxidase (HRP)-conjugated
anti-mouse IgG and anti-rabbit IgG secondary antibody (Amersham
Biosciences, Piscataway, NJ, USA) in blocking buffer. Finally, the
blots were washed three times with TBST buffer and visualized via
enzyme-linked chemiluminescence using the electrochemiluminescence
(ECL) kit (Applygen Technologies Inc.) (23). The results of western blotting were
semi-quantitatively analyzed by ImageJ software (NIH, Bethesda, MD,
USA).
Protein levels were normalized with GAPDH, and the
levels of phospho-AKT immunoreactivity were normalized to the total
AKT immunoreactivity. The primary antibody specific for the EBP50
was purchased from BD Biosciences (San Jose, CA, USA), anti-HA was
from MBL (Nagoya, Japan), anti-Flag antibody was from
Sigma-Aldrich. Other primary antibodies specific for GAPDH, PTEN,
phospho-AKT (Ser473), total AKT were all bought from Cell Signaling
Technology (Beverly, MA, USA).
GST pull-down assay
GST fusion proteins were purified from bacteria
using glutathione-sepharose 4B beads (Sigma-Aldrich) according to
the manufacturer's protocol. The GST pull-down assay was performed
as described previously (24).
Briefly, equal amounts of GST or GST-PTEN-CT (amino acid from 374
to 403, WT or V403A mutant-the last amino acid V403 mutated to A)
fusion protein beads were incubated with equal amounts of cell
lysates. After incubation at 4°C for 2 h, the beads were washed
with ice-cold wash buffer. Proteins were then eluted with SDS
sample buffer, and detected with western blotting.
Co-immunoprecipitation assay
Co-immunoprecipitation (Co-IP) was performed as
described (25). Briefly, the
rabbit kidney tissues were homogenized in ice-cold lysis buffer (10
mmol/l HEPES, 50 mmol/l NaCl, 5 mmol/l EDTA, 1 mmol/l benzamidine,
0.5% Triton X-100, pH 7.4). Lysates were solubilized and clarified.
Supernatants were incubated with anti-PTEN antibody (Cell Signaling
Technology), prebounded with protein A&G beads (Calbiochem, San
Diego, CA, USA). After washing with an ice-cold lysis buffer three
times, the immunoprecipitated proteins were eluted from the beads
with SDS sample loading buffer. The eluted samples were then
analyzed by western blotting. Anti-PTEN and anti-EGFR antibody were
used in this study.
Statistical analyses
All data are presented as means ± SD and statistical
significance was analyzed by ANOVA. Differences were considered
significant at P<0.05.
Results
PTEN forms a complex with EGFR in
tissues
In order to verify that PTEN formed a complex with
EGFR, we chose rabbit kidney tissues which express high level of
PTEN, EGFR and EBP50 to perform Co-IP study. Solubilized lysates
from rabbit kidney tissues were incubated with IgG or PTEN antibody
linked to protein A/G-agarose beads. Co-precipitated EGFR was
detected by western blotting with anti-EGFR antibody.
Co-immunoprecipitation of EGFR with PTEN in rabbit kidney tissues
was detected, whereas no co-immunoprecipitation of IgG with PTEN
was observed (Fig. 1). Co-IP assay
result of tissue showed that PTEN could co-immunoprecipate with
EGFR and provided evidence of a physical complex between PTEN and
EGFR in tissues.
EBP50 protein mediates the formation of
the PTEN/EGFR complex
To further clarify whether EGFR forms a complex with
PTEN via their direct interaction, or via adaptor protein EBP50, we
detected the formation of PTEN/EGFR complex in the absence of
EBP50. COS-7 cells were transfected with the pSuper.puro-EBP50
shRNA plasmid or the control plasmid, respectively. The EBP50
stable knockdown cell line (shEBP) and its control cell line
(shLuc) were generated by puromycin screening. Verification of
protein knockdown was determined by western blot analysis as shown
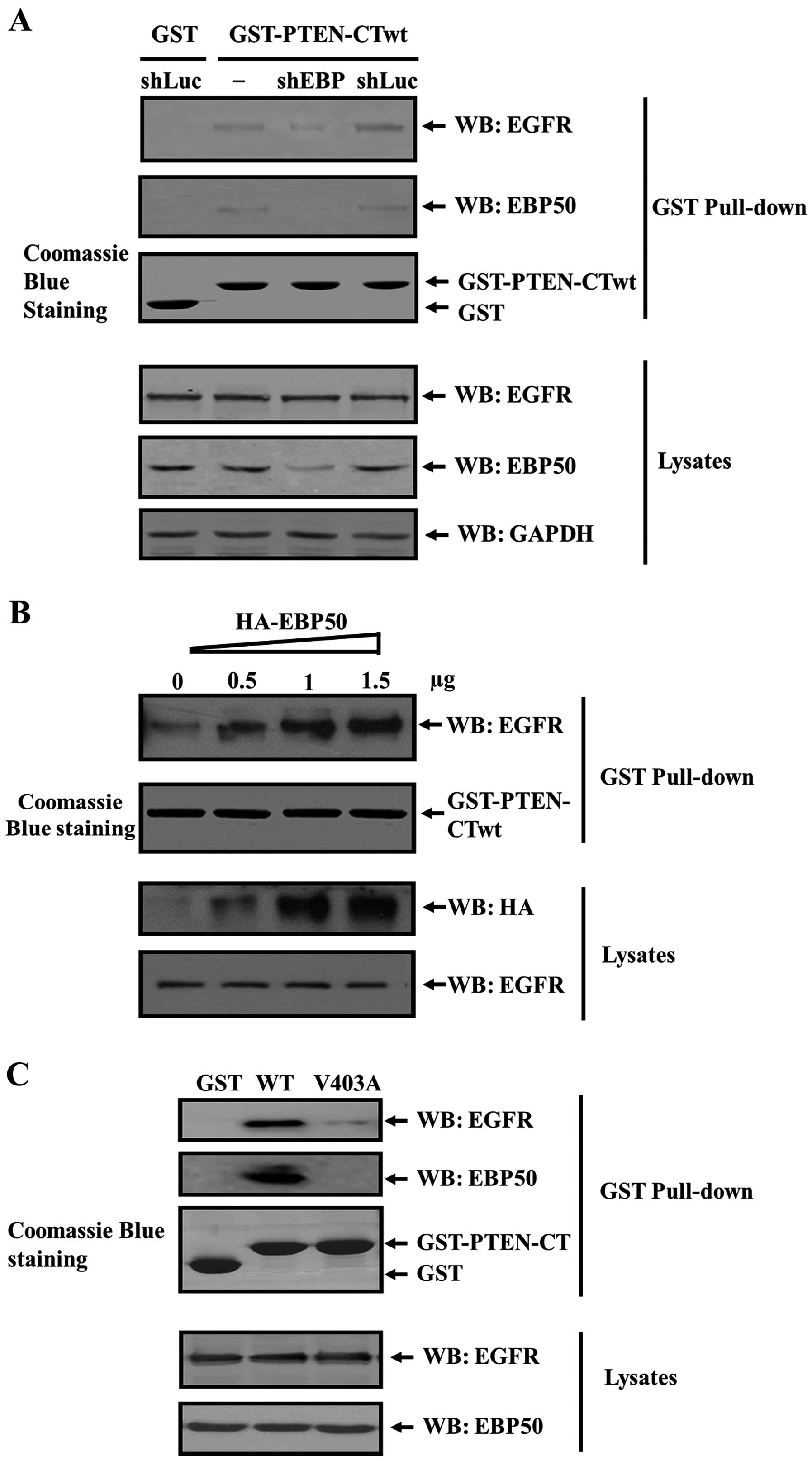
in Fig. 2A. In shLuc cells, EBP50
expression level was the same as that in parental cells. In shEBP
cells, EBP50 expression was stably knocked down up to 67% compared
to that of its parental cells. Subsequently, these cell lysates
were collected and subjected to GST pull-down assay. As shown in
Fig. 2A, EGFR and EBP50 signals
were detected from the GST-PTEN-CT pull-down complex in COS-7
parental cells and shLuc cells, but not in shEBP cells, indicating
PTEN/EGFR complex formation was mediated by EBP50 and the knockdown
of EBP50 disrupted the ternary complex formation.
EBP50 knockdown disrupted the formation of the
PTEN/EGFR complex, preliminarily suggesting that EBP50 can act as
an adaptor to assemble the PTEN/EGFR complex. Thus, it is possible
that the increase of EBP50 expression level will lead to an
increase in the complex amount. We further detected whether
PTEN/EGFR complex was facilitated in EBP50-concentration dependent
manner. We transiently transfected increasing dose of
pBK-CMV-HA-EBP50 expression plasmid (0, 0.5, 1 and 1.5 µg,
respectively) into COS-7 cells, then collected the lysates of these
cells to perform GST pull-down experiment and to detect EGFR in the
GST pull-down fraction. Results showed that in cell lysates the
expression level of exogenous EBP50 protein increased. Whereas,
EGFR expression level was not influenced. When these cell lysates
were pulled down by GST-PTEN-CTwt fusion protein, with the increase
of EBP50 expression level, the amount of EGFR pulled down by
GST-PTEN-CTwt increased accordingly (Fig. 2B), further verifying that EBP50
mediated the formation of PTEN/EGFR complex.
EBP50 bound with PTEN and EGFR via their PDZ binding
motifs. When PDZ binding motif of PTEN or EGFR is mutated (PTEN
V403A, EGFR L1043/1063F) and can not bind with EBP50, the PTEN/EGFR
complex may not form. To confirm this hypothesis, we expressed
GST-PTEN-CT wild-type (wt) and V403A mutant fusion proteins,
respectively, and used them to pull down COS-7 cell lysates. As
shown in Fig. 2C, GST-PTEN-CT-wt
can pull down EBP50 and EGFR from COS-7 lysates, whereas GST and
GST-PTEN-CT-mt (V403A) can not. This suggested that EGFR did not
associate with PTEN when EBP50 could not bind with PTEN, indicating
that EBP50 specifically mediated the formation of PTEN/EGFR
complex.
Taken together, EBP50 mediated the PTEN/EGFR complex
formation and the adaptor role of EBP50 as PTEN/EGFR complex is
specific.
PTEN binding with EGFR regulates EGFR/AKT
signaling
PTEN/EBP50/PDGFR complex regulated PDGF-induced AKT
signaling (14), and the
PTEN/EBP50/EGFR complex may also regulate EGF-induced AKT
signaling. We next assessed the regulatory roles of EBP50 and PTEN
in EGF-induced AKT activation. Flag control vector, flag-PTEN-WT,
flag-PTEN-V403A mutant was transiently transfected into COS-7
cells, respectively, then EGF stimulation was performed for 30 min
to observe the EGF-induced AKT activation level in these
transfected cells. In PTEN-WT transfected cells, AKT activity by
EGF stimulation was lower than that in vector control cells (only
34% of that in vector control cells). The AKT activation level in
PTEN-V403A transfected cells was relatively higher than that in
PTEN-WT transfected cells (60% of that in vector control cells,
P<0.05, Fig. 3A).
Interestingly, similar result was revealed in U373
cells which have no endogenous PTEN expression. In PTEN-WT
transfected U373 cells, AKT activity by EGF stimulation was lower
than that in vector control cells (only 23% of that in vector
control cells), but in PTEN-V403A transfected U373 cells AKT
activation level was relatively higher than that in PTEN-WT
transfected U373 cells (54% of that in vector control cells)
(P<0.05, Fig. 3B). As PTEN-V403A
mutant fails to interact with EBP50, it is unable to mediate the
formation of PTEN/EBP50/EGFR trimer. This result indicated that
more active AKT in PTEN-V403A transfected cells compared with
PTEN-WT transfected cells may result from the failure of
PTEN/EBP50/EGFR trimer formation, and consequent lack of ability of
PTEN in inhibiting EGF-induced AKT pathway.
EGFR L1043/1063F mutant can not bind with EBP50
(21). To further confirm the role
of the EBP50-mediated PTEN/EGFR complex, flag control vector,
flag-EGFR-WT, flag-EGFR-L1043/1063F mutant which can not bind to
EBP50 was transiently transfected into COS-7 cells, respectively.
Then EGF stimulation was performed for 30 min to observe the
activation level of AKT in these transfected cells. Results showed
that, in EGFR-WT transfected cells, AKT activation level by EGF
stimulation was higher than that in vector control cells (1.43-fold
over that in vector control cells). However, AKT activation in
EGFR-L1043/1063F transfected cells was relatively higher than that
in vector control cells, even higher than that in EGFR-WT
transfected cells (2.22-fold over that in vector control cells and
1.56-fold over that in EGFR-WT transfected cells, respectively)
(P<0.05, Fig. 4A). As
EGFR-L1043/1063F mutant can not interact with EBP50, it is unable
to mediate the formation of PTEN/EBP50/EGFR trimer. This result
further indicated that it was the failure of the trimer formation
that caused the failure of PTEN in inhibiting EGFR-AKT pathway and
more active AKT in EGFR-L1043/1063F mutant transfected cells than
EGFR-WT transfected cells.
Consistent with these results, disruption of this
complex led to an increase of EGF-induced AKT activation level.
EBP50 siRNA was used to knock down the expression of EBP50 in COS-7
cells, its EBP50 expression was knocked down by ~52%, and its AKT
activation level was 1.57- and 1.62-fold over that in scrambled
siRNA transfected cells at 15 and 30 min of EGF stimulation,
respectively (P<0.05, Fig. 4B),
revealing that EBP50 knockdown led to stronger AKT activation.
Thus, these results consistently demonstrate that the ternary
complex between PTEN, EBP50 and EGFR leads to a specific inhibition
role for EGF-induced AKT activation.
Altogether, these results clearly reveal that
EBP50-mediated PTEN/EGFR complex can inhibit EGF-induced EGFR/AKT
pathway activation more effectively.
Discussion
In this study, we found that EBP50 mediated
EGFR/PTEN super-macromolecular complexation and inhibited the
activation of EGF-induced AKT signaling pathway. Mutating either
EGFR (L1043/1063F) or PTEN (V403A) to abolish its binding with
EBP50, severely interrupted the formation of PTEN/EGFR complex, and
knockdown of EBP50 expression in cells also abolished the PTEN/EGFR
complex. Overexpressing EGFR (L1043/1063F) or PTEN (V403A) mutants,
which can not form PTEN/EGFR complex, in cells enhanced the
phosphorylation level of EGF-induced AKT. Consistently, EBP50
knockdown in cells also enhanced the phosphorylation level of
EGF-induced AKT. These results demonstrated that EBP50 as a
scaffold organized the assembly of PTEN/EGFR complex and mediated
the inhibition of AKT activation induced by EGF. This finding
reveals a new macromolecular signaling complex involving in EGFR
and helps to better understand the regulatory mechanism of EGFR
signaling pathway.
PTEN phosphatase domain is located in its N-terminal
(NT) and PTEN-NT missense mutant lost its phosphatase activity
(26,27). Interestingly, PTEN C-terminal (CT,
codons 212–403) missense mutants were also reported to have
relatively low phosphatase activity (28–31).
In this study we found that PTEN-CT V403A mutant also showed low
phosphatase activity for pAKT (Fig.
3). This phenomenon remains a mystery. We found PTEN-CT was
very important in binding with EBP50 and forming a complex with
EGFR. This complex could attenuate EGF-induced AKT activity,
providing the additional mechanism that PTEN CT mutant weakens its
ability to suppress EGF-induced AKT activation.
In clinical samples, some EGFR mutation sites [T790M
(32), L858R (33), L861Q (34)] were not in the autophosphorylation
site of EGFR, but their signaling activation ability was stronger
than that of EGFR WT. Some EGFR single nucleotide polymorphisms
[SNPs, R962G, R977C, H988P (35)]
were not in the autophosphorylation site of EGFR, but their
signaling activation ability was also stronger than that of EGFR
WT. Its molecular mechanism remains unknown. These EGFR mutations
or SNPs are close to the EBP50 interacting site (1037–1065), it is
possible that the stronger EGFR signaling activation results from
the failure in forming the EGFR/EBP50/PTEN complex and further
failure in inhibiting EGF-induced AKT signaling. In this study,
EGFR mutant (L1043/1063F) expression also triggered stronger
EGFR-mediated AKT signaling. Its mechanism was involved in failure
in forming the EGFR/EBP50/PTEN complex, supporting the important
clinical significance of EBP50 binding in EGFR signaling
regulation. However, this needs to be further explored.
As an adaptor protein, EBP50 plays an important role
in the formation of the EGFR/EBP50/PTEN complex. The change of
EBP50 expression level possibly influence the formation of the
complex, further influencing the activation of AKT pathway. Thus,
we performed data mining from the public database NCBIs Gene
Expression omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) Profile (GSE6344
data from clear-cell renal cell carcinoma cases) and selected
samples with similar EGFR, PTEN and AKT gene
expression levels between cancer and paracancer tissues (P>0.05
according to paired-sample t-test paracancer samples vs. cancer
samples. EGFR 201.64±68.23 vs. 199.13±67.54; PTEN 24.93±21.99 vs.
22.56±25.71, AKT 592.72±87.42 vs. 731.94±153.48). We then examined
the correlation between expression levels of EBP50 and gene sets
involved in EGFR and AKT pathway in clinical samples by Gene set
enrichment analysis (GSEA; http://www.broad.Mit.edu/gsea). The expression level
of a priory defined positive EGFR gene set (GNF2_EGFR) and
AKT pathway gene set (PID_PI3KCI_AKT_pathway and
KEGG_mTOR_Signaling_pathway, c2. v4.0 from the Molecular Signatures
Database-MsigDB, http://www.broad.mit.edu/gsea/msigdb/index.jsp) were
used as an indicator of the activation of the EGFR pathway and AKT
pathway, respectively. GSEA result showed that both their EGFR gene
set and AKT gene set were enriched in EBP50 lower expression
patients, respectively, suggesting that EBP50 expression negatively
contributed to EGFR and AKT gene set expression in these patients
(FDR=0.002, P=0.001 and FDR=0.03, P=0.02, respectively). GSEA
result partly verified EBP50 retarded EGF-induced AKT signaling
from clinical data, also supporting that EGFR/EBP50/PTEN complex
plays an important regulatory role in EGFR and AKT signaling
pathway.
In addition, in some cervical clear cell carcinoma
cases, AKT phosphorylation level increased (36). Interestingly, the expression level
of EGFR which can increase AKT phosphorylation level did not
significantly increase (from the human protein atlas result) or
EGFR was even expressed in low level (data analysis results of
Pubmed GEO Profile cervical carcinoma data GDS3233), reminding that
the increase of AKT phosphorylation level may not completely result
from the increase of EGFR expression level. EBP50 expression level
was significantly downregulated (from the Human Protein Atlas
result and GDS3233 data, paracancer samples 10.06±0.65 vs. cancer
samples 9.07±0.88, P<0.01, independent-samples t-test). Because
this study demonstrated that EBP50 mediated the formation of
EGFR/PTEN complex, it is possible that decreased EBP50 expression
in this type of tumor may explain the pathogenesis and further
elucidate the importance of the PTEN-EBP50 connection for cancer
progression. However, this needs to be further experimentally
verified.
EBP50 may behave either as a tumor suppressor, when
it is localized at the plasma membrane (PM), or as an oncogenic
protein, when it is at the cytoplasm and nuclei (37,38).
Its molecular mechanism remains unclear. Current results showed
that only when EBP50 bound with EGFR localized in PM, it can
recruit PTEN and form a complex with EGFR, then regulate
EGF-induced AKT phosphorylation. When EBP50 did not bind with EGFR
localized at PM, it can not mediate the EGFR/PTEN complex formation
and regulate EGF-induced AKT signaling. Binding with EGFR in PM may
be one of the mechanisms for EBP50 to be a tumor suppressor.
In summary, this study clarified the nature of
EGFR/AKT pathway regulation by PTEN under the mediation of EBP50,
and demonstrated an additional level of extrinsic protein
regulation through binding partner interactions. EBP50 and PTEN
proteins suppress EGF-induced AKT signaling and points to a helper
role for EBP50 proteins in PTEN tumor suppressor activity.
Acknowledgments
This study was supported by the National Natural
Science Foundation of the People's Republic of China (no. 30900247
and 81372739), the Importation and Development of High-Caliber
Talents Project of Beijing Municipal Institutions
(CIT&TCD201304187) and Beijing Municipal Science and Technology
Commission (no. Z151100001615039).
Abbreviations:
|
EGFR
|
epidermal growth factor receptor
|
|
PTEN
|
phosphatase and tensin homolog deleted
on chromosome ten
|
|
EBP50
|
ezrin-radixin-moesin-binding
phosphoprotein-50
|
|
PDGFR
|
platelet-derived growth factor
receptor
|
|
PI3K/AKT
|
phosphoinositide 3-kinase/AKT
|
|
MAPK
|
mitogen-activated protein kinase
|
|
STAT
|
signal transducer and activator of
transcription
|
References
|
1
|
Safdari Y, Khalili M, Ebrahimzadeh MA,
Yazdani Y and Farajnia S: Natural inhibitors of PI3K/AKT signaling
in breast cancer: Emphasis on newly-discovered molecular mechanisms
of action. Pharmacol Res. 93:1–10. 2015. View Article : Google Scholar
|
|
2
|
Jones S and Rappoport JZ: Interdependent
epidermal growth factor receptor signalling and trafficking. Int J
Biochem Cell Biol. 51:23–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pan D and Lin X: Epithelial growth factor
receptor-activated nuclear factor κB signaling and its role in
epithelial growth factor receptor-associated tumors. Cancer J.
19:461–467. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Han W and Lo HW: Landscape of EGFR
signaling network in human cancers: Biology and therapeutic
response in relation to receptor subcellular locations. Cancer
Lett. 318:124–134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu R, Gu J, Jiang P, Zheng Y, Liu X,
Jiang X, Huang E, Xiong S, Xu F, Liu G, et al: DNMT1-microRNA126
epigenetic circuit contributes to esophageal squamous cell
carcinoma growth via ADAM9-EGFR-AKT signaling. Clin Cancer Res.
21:854–863. 2015. View Article : Google Scholar
|
|
6
|
Khabele D, Kabir SM, Dong Y, Lee E, Rice
VM and Son DS: Preferential effect of akt2-dependent signaling on
the cellular viability of ovarian cancer cells in response to EGF.
J Cancer. 5:670–678. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu G, Jiang C, Li D, Wang R and Wang W:
miRNA-34a inhibits EGFR-signaling-dependent MMP7 activation in
gastric cancer. Tumour Biol. 35:9801–9806. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pei J, Lou Y, Zhong R and Han B: MMP9
activation triggered by epidermal growth factor induced Foxo1
nuclear exclusion in non-small cell lung cancer. Tumour Biol.
35:6673–6678. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang XJ, Feng CW and Li M: ADAM17 mediates
hypoxia-induced drug resistance in hepatocellular carcinoma cells
through activation of EGFR/PI3K/Akt pathway. Mol Cell Biochem.
380:57–66. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu W, Ren H, Ren J, Yin T, Hu B, Xie S,
Dai Y, Wu W, Xiao Z, Yang X, et al: The role of
EGFR/PI3K/Akt/cyclinD1 signaling pathway in acquired middle ear
cholesteatoma. Mediators Inflamm. 2013:6512072013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tiganis T, Kemp BE and Tonks NK: The
protein-tyrosine phosphatase TCPTP regulates epidermal growth
factor receptor-mediated and phosphatidylinositol
3-kinase-dependent signaling. J Biol Chem. 274:27768–27775. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Scharl M, Rudenko I and McCole DF: Loss of
protein tyrosine phosphatase N2 potentiates epidermal growth factor
suppression of intestinal epithelial chloride secretion. Am J
Physiol Gastrointest Liver Physiol. 299:G935–G945. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Omerovic J, Clague MJ and Prior IA:
Phosphatome profiling reveals PTPN2, PTPRJ and PTEN as potent
negative regulators of PKB/Akt activation in Ras-mutated cancer
cells. Biochem J. 426:65–72. 2010. View Article : Google Scholar
|
|
14
|
Takahashi Y, Morales FC, Kreimann EL and
Georgescu MM: PTEN tumor suppressor associates with NHERF proteins
to attenuate PDGF receptor signaling. EMBO J. 25:910–920. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maudsley S, Zamah AM, Rahman N, Blitzer
JT, Luttrell LM, Lefkowitz RJ and Hall RA: Platelet-derived growth
factor receptor association with Na(+)/H(+) exchanger regulatory
factor potentiates receptor activity. Mol Cell Biol. 20:8352–8363.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang L, Wang Y, Chen P, Hu J, Xiong Y,
Feng D, Liu H, Zhang H, Yang H and He J: Na(+)/H(+) exchanger
regulatory factor 1 (NHERF1) is required for the
estradiol-dependent increase of phosphatase and tensin homolog
(PTEN) protein expression. Endocrinology. 152:4537–4549. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pan Y, Weinman EJ and Dai JL:
Na+/H+ exchanger regulatory factor 1 inhibits
platelet-derived growth factor signaling in breast cancer cells.
Breast Cancer Res. 10:R52008. View
Article : Google Scholar
|
|
18
|
Cheng S, Li Y, Yang Y, Feng D, Yang L, Ma
Q, Zheng S, Meng R, Wang S, Wang S, et al: Breast cancer-derived
K172N, D301V mutations abolish Na+/H+
exchanger regulatory factor 1 inhibition of platelet-derived growth
factor receptor signaling. FEBS Lett. 587:3289–3295. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rodriguez S and Huynh-Do U: Phosphatase
and tensin homolog regulates stability and activity of EphB1
receptor. FASEB J. 27:632–644. 2013. View Article : Google Scholar
|
|
20
|
Yao W, Feng D, Bian W, Yang L, Li Y, Yang
Z, Xiong Y, Zheng J, Zhai R and He J: EBP50 inhibits EGF-induced
breast cancer cell proliferation by blocking EGFR phosphorylation.
Amino Acids. 43:2027–2035. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lazar CS, Cresson CM, Lauffenburger DA and
Gill GN: The Na+/H+ exchanger regulatory
factor stabilizes epidermal growth factor receptors at the cell
surface. Mol Biol Cell. 15:5470–5480. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng J, Shen H, Xiong Y, Yang X and He J:
The beta1-adrenergic receptor mediates extracellular
signal-regulated kinase activation via Galphas. Amino Acids.
38:75–84. 2010. View Article : Google Scholar
|
|
23
|
Zheng JF, Sun LC, Liu H, Huang Y, Li Y and
He J: EBP50 exerts tumor suppressor activity by promoting cell
apoptosis and retarding extracellular signal-regulated kinase
activity. Amino Acids. 38:1261–1268. 2010. View Article : Google Scholar
|
|
24
|
Sun C, Zheng J, Cheng S, Feng D and He J:
EBP50 phosphorylation by Cdc2/cyclin B kinase affects actin
cytoskeleton reorganization and regulates functions of human breast
cancer cell line MDA-MB-231. Mol Cells. 36:47–54. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang X, Zheng J, Xiong Y, Shen H, Sun L,
Huang Y, Sun C, Li Y and He J: Beta-2 adrenergic receptor mediated
ERK activation is regulated by interaction with MAGI-3. FEBS Lett.
584:2207–2212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He X, Arrotta N, Radhakrishnan D, Wang Y,
Romigh T and Eng C: Cowden syndrome-related mutations in PTEN
associate with enhanced proteasome activity. Cancer Res.
73:3029–3040. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu J, Li Z, Wang J, Chen H and Fang JY:
Combined PTEN mutation and protein expression associate with
overall and disease-free survival of glioblastoma patients. Transl
oncol. 7:196–205.e1. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang SJ, Endo S, Ichikawa T, Yoshimura J,
Onda K, Tanaka R, Washiyama K and Kumanishi T: Rare-type mutations
of MMAC1 tumor suppressor gene in human glioma cell lines and their
tumors of origin. Jpn J Cancer Res. 90:934–941. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Valiente M, Andrés-Pons A, Gomar B, Torres
J, Gil A, Tapparel C, Antonarakis SE and Pulido R: Binding of PTEN
to specific PDZ domains contributes to PTEN protein stability and
phosphory-lation by microtubule-associated serine/threonine
kinases. J Biol Chem. 280:28936–28943. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu D, Yao Y, Jiang X, Lu L and Dai W:
Regulation of PTEN stability and activity by Plk3. J Biol Chem.
285:39935–39942. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maxwell GL, Risinger JI, Gumbs C, Shaw H,
Bentley RC, Barrett JC, Berchuck A and Futreal PA: Mutation of the
PTEN tumor suppressor gene in endometrial hyperplasias. Cancer Res.
58:2500–2503. 1998.PubMed/NCBI
|
|
32
|
Barton S, Starling N and Swanton C:
Predictive molecular markers of response to epidermal growth factor
receptor (EGFR) family-targeted therapies. Curr Cancer Drug
Targets. 10:799–812. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Suzuki T, Fujii A, Ohya J, Amano Y, Kitano
Y, Abe D and Nakamura H: Pharmacological characterization of MP-412
(AV-412), a dual epidermal growth factor receptor and ErbB2
tyrosine kinase inhibitor. Cancer Sci. 98:1977–1984. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang S, Qu S, Perez-Tores M, Sawai A,
Rosen N, Solit DB and Arteaga CL: Association with HSP90 inhibits
Cbl-mediated down-regulation of mutant epidermal growth factor
receptors. Cancer Res. 66:6990–6997. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Choura M, Frikha F, Kharrat N, Aifa S and
Rebaï A: Investigating the function of three non-synonymous SNPs in
EGFR gene: Structural modelling and association with breast cancer.
Protein J. 29:50–54. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ueno S, Sudo T, Oka N, Wakahashi S,
Yamaguchi S, Fujiwara K, Mikami Y and Nishimura R: Absence of human
papillomavirus infection and activation of PI3K-AKT pathway in
cervical clear cell carcinoma. Int J Gynecol Cancer. 23:1084–1091.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Georgescu MM, Morales FC, Molina JR and
Hayashi Y: Roles of NHERF1/EBP50 in cancer. Curr Mol Med.
8:459–468. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shibata T, Chuma M, Kokubu A, Sakamoto M
and Hirohashi S: EBP50, a beta-catenin-associating protein,
enhances Wnt signaling and is over-expressed in hepatocellular
carcinoma. Hepatology. 38:178–186. 2003. View Article : Google Scholar : PubMed/NCBI
|