Introduction
Thioredoxin (Trx) and glutathione (GSH) are major
antioxidant systems in the cells to defend excess reactive oxygen
species (ROS) production. Trx system consists of Trx and
nicotinamide adenine dinucleotide phosphate (NADPH)-dependent Trx
reductase (TrxR) (1). Trx, having
two active sites in cysteine residue, exists as a dithiol, reduced
form. When Trx is oxidized, it is reduced by TrxR (1). GSH is a non-protein antioxidant and
stabilizes the oxidized molecules by supplying electron. Trx and
GSH systems control not only redox status but also affect many
cellular events such as proliferation and apoptosis (2–4).
Especially, TrxR1 is overexpressed in breast and oral cancer
patients (5,6). It has been reported that the
inhibition of TrxR increases the sensitivity of cancer cells to
radiotherapy and anticancer drugs in melanoma, colon and breast
cancers (7–9). Therefore, the regulation of Trx system
can be a promising target for cancer therapy (10).
Auranofin, as a TrxR inhibitor, is used for the
treatment of rheumatoid arthritis. Originally, this agent was
considered as anti-inflammatory drug (11). However, recently many studies
demonstrate that auranofin has an anticancer effect in leukemia and
ovarian cancer cells (12,13). In addition, It has been suggested
that auranofin induces FOXO3 activation, ROS accumulation, DNA
damage and ERK inactivation in cancer cells (13,14).
Mesothelioma is a rare tumor mainly derived from the pleura of lung
and it has a poor prognosis (15).
Although it is reported that TrxR1 is overexpressed in mesothelioma
cells (16), little is known about
the anti-growth effect of auranofin in mesothelioma cells.
In the present study, we investigated the effects of
auranofin on cell proliferation and death in patient-derived human
mesothelioma cells in relation to ROS and GSH levels.
Materials and methods
Cell culture
Human mesothelial cells (HM69 and HM72) and human
mesothelioma cells (ADA, CON, Hmeso, Mill, Phi, REN and ROB) were
obtained from Queen's Medical Center (Honolulu, HI, USA). These
cells were cultured in Ham's F-12 media containing 10% fetal bovine
serum (FBS) and 1% penicillin-streptomycin (both from Gibco BRL,
Grand Island, NY, USA). Mesothelial and mesothelioma cells were
maintained in incubator containing 5% CO2 at 37°C. Cells
were grown in 100 mm plastic cell culture dishes (BD Falcon,
Franklin Lakes, NJ, USA) and harvested with a trypsin-EDTA (Gibco
BRL).
Reagents
Auranofin purchased from Santa Cruz Biotechnology
(Santa Cruz, CA, USA) was dissolved in dimethyl sulfoxide (DMSO;
Sigma-Aldrich Chemical Co., St. Louis, MO, USA) at 10 mM as a stock
solution. The pan-caspase inhibitor (Z-VAD-FMK;
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone) was obtained from
R&D Systems, Inc. (Minneapolis, MN, USA) and were dissolved in
DMSO at 10 mM. NecroX-2 and necrostatin-1 from Enzo Life Science
(Plymouth Meeting, PA, USA) were dissolved in DMSO at 1 and 50 mM,
respectively. NAC and BSO obtained from Sigma-Aldrich Chemical Co.
were dissolved in 20 mM HEPES (pH 7.0) and water at 100 mM,
respectively. Cells were pretreated with 15 μM Z-VAD, 1
μM NecroX-2, 50 μM necrostatin-1, 2 mM NAC or 10
μM BSO for 1 h prior to auranofin treatment.
Western blot analysis
The protein expression levels were evaluated by
western blot analysis. In brief, 1×106 cells in 60 mm
culture dish (BD Falcon) were incubated with or without 3 μM
auranofin for 24 h. Then cells were washed with phosphate-buffered
saline (PBS) and added in 4 volumes of protein extract buffer (Life
Technologies, Carlsbad, CA, USA). The concentrations of protein
were determined using the Bradford method. A total of 30 μg
total proteins were resolved by 4–20% SDS-PAGE gels, and then
transferred to Immobilon-P PVDF membranes (Millipore, Billerica,
MA, USA) by electroblotting. Then membranes were probed with
anti-PARP and anti-c-PARP (Cell signaling Technology, Danvers, MA,
USA) and anti-TrxR1, anti-GAPDH and anti-β-actin (Santa Cruz
Biotechnology). Membrane was incubated with fluorescence-conjugated
secondary antibodies. Bands were visualized by using a LI-COR
Odyssey Imager (LI-COR Biosciences, Lincoln, NE, USA).
Cell proliferation assay
The effect of auranofin on proliferation in
mesothelioma cells was determined by CellTiter 96®
AQueous Non-Radioactive Cell Proliferation Assay kit (Promega,
Madison, WI, USA). In brief, 5×103 cells in 96-well
microtiter plate (BD Falcon) were incubated with the indicated
concentrations of auranofin with or without NAC or BSO for 24 h.
Then, 20 μl of 3-(4,5-dimethylthazol-2-yl)-5-(3-carboxy
methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) and phenazine
methosulfate (PMS) mixture was added to each well in 96-well
plates. The plates were incubated for 3 h at 37°C. The optical
density was measured at 490 nm using a microplate reader (VersaMax
plate reader; Molecular Devices, Sunnyvale, CA, USA).
Sub G-1 analysis
Sub-G1 analysis was determined by propidium iodide
(PI; Sigma-Aldrich Chemical Co.; Ex/Em=488/617 nm) staining.
Briefly, 1×106 cells in 60 mm culture dish (BD Falcon)
were incubated with the indicated concentrations of auranofin with
or without Z-VAD, NecroX-2 or necrostatin-1 for 24 h. Cells were
washed with PBS and then incubated with 10 μg/ml PI with
RNase at 37°C for 30 min. Sub-G1 DNA content cells were analyzed
with an Accuri C6 flow cytometer (BD Sciences, Franklin Lakes, NJ,
USA).
Annexin V/PI staining
Apoptosis was detected by staining cells with
Annexin V-fluorescein isothiocyanate (FITC; Life Technologies;
Ex/Em=488/519 nm) and PI (Sigma-Aldrich Chemical Co.). Briefly,
1×106 cells in 60 mm culture dish (BD Falcon) were
incubated with the indicated concentrations of auranofin with or
without Z-VAD, NecroX-2, necrostatin-1, NAC or BSO for 24 h. Then
cells were washed twice with cold PBS and added 500 μl of
binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM
CaCl2) at a concentration of 1×106 cells/ml.
Five microliters of Annexin V-FITC and PI were added to these
cells, which were analyzed with Accuri C6 flow cytometer (BD
Sciences).
Lactate dehydrogenase (LDH) release
assay
Necrosis in cells was evaluated by LDH kit
(Sigma-Aldrich Chemical Co.) Briefly, 1×106 cells in 60
mm culture dish (BD Falcon) were incubated with the indicated
concentration of auranofin with or without NAC or BSO for 24 h.
After treatment, the cell culture media were collected and
centrifuged for 5 min at 1,500 rpm. A total of 50 μl of the
media supernatant was added to a fresh 96-well plate (SPL Life
Sciences, Pocheon, Gyeonggi-do, Korea) with LDH assay reagent and
then incubated at room temperature for 30 min. The absorbance
values were measured at 490 nm using a microplate reader (Synergy™
2; BioTek® Instruments Inc., Winooski, VT, USA). LDH
release was expressed as the percentage of extracellular LDH
activity compared with the control cells.
Detection of intracellular ROS
levels
Intracellular ROS such as
H2O2, •OH and ONOO•
were detected by an oxidation-sensitive fluorescent probe dye,
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA,
Life Technologies; Ex/Em=495/529 nm). As H2DCFDA is
poorly selective for O2•−, dihydroethidium
(DHE, Life Technologies; Ex/Em=518/605 nm), which is highly
selective for O2•−, was used for its
detection. Briefly, 1×106 cells in 60 mm culture dish
(BD Falcon) were incubated with the indicated concentrations of
auranofin with or without NAC or BSO for 24 h. The cells were
washed in PBS and incubated with 20 μM H2DCFDA
and DHE at 37°C for 30 min. DCF and DHE fluorescences were detected
by using Accuri C6 flow cytometer (BD Sciences).
Measurement of intracellular GSH
level
Cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA, Ex/Em=522/595 nm;
Life Technologies). In brief, 1×106 cells were incubated
in a 60 mm culture dishes (BD Falcon) with 3 μM auranofin
with or without NAC or BSO for 24 h. Cells were then washed with
PBS and incubated with 5 μM CMFDA at 37°C for 30 min. CMF
fluorescence intensity was determined using Accuri C6 flow
cytometer (BD Sciences). Negative CMF staining (GSH-depletion) of
cells is expressed as the percentage of (−) CMF cells.
Statistical analysis
The results represent the mean of at least three
independent experiments (mean ± SD). Data were analyzed using
Instat software (GraphPad Prism4, San Diego, CA, USA). The
Student's t-test or one-way analysis of variance (ANOVA) with post
hoc analysis using Tukey's multiple comparison test was used for
parametric data. P<0.05 was considered to indicate a
statistically significant difference.
Results
Auranofin inhibits proliferation and
induces the death of mesothelioma cells
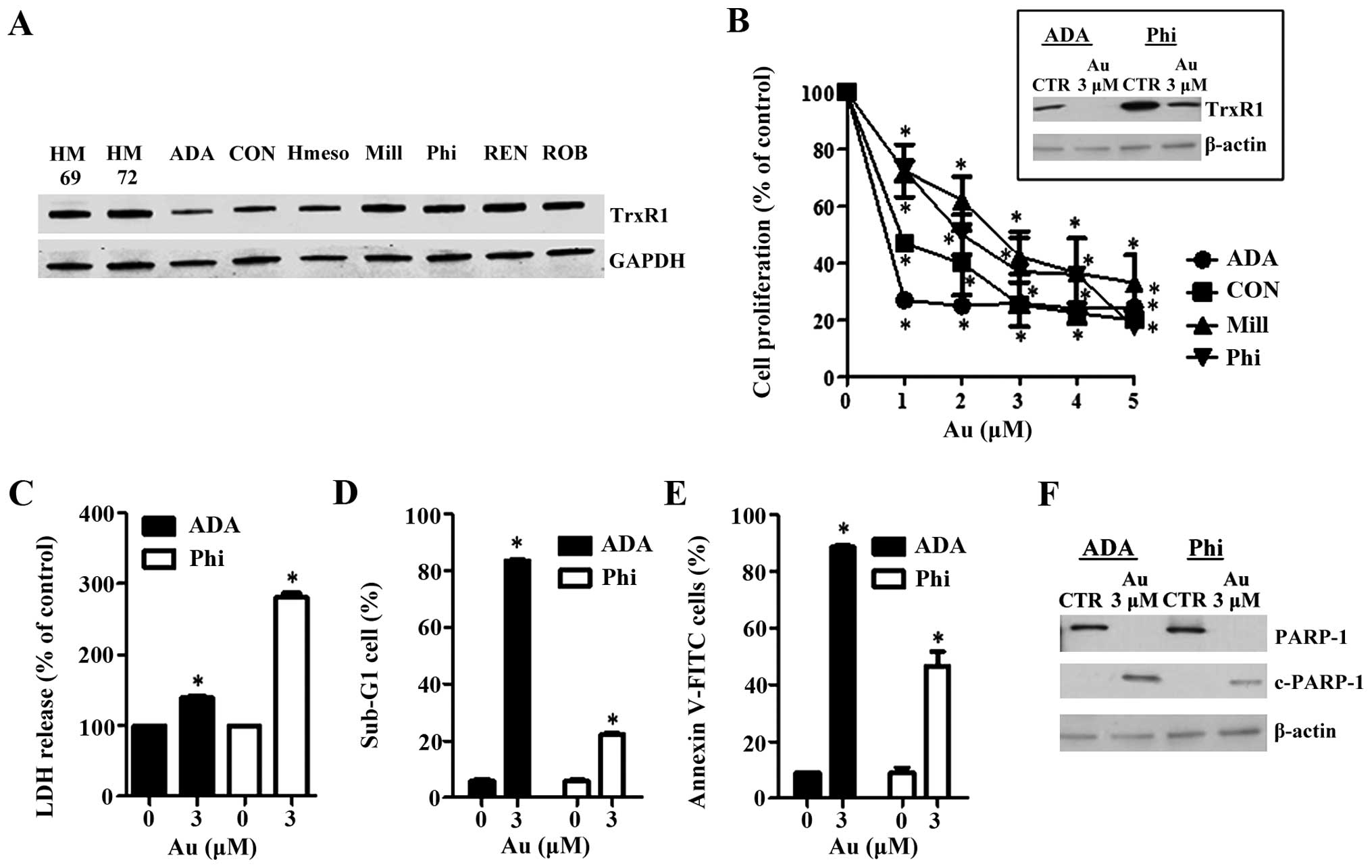
Firstly, we observed the protein expression levels
of TrxR1 in human mesothelial and mesothelioma cells. As a result,
there was no difference of TrxR1 expression in either mesothelial
or mesothelioma cells (Fig. 1A).
Instead, the levels of TrxR1 in ADA, CON and Hmeso were lower than
those in other mesothelioma cells (Fig.
1A). Treatment with auranofin attenuated the proliferation of
mesothelioma cells in a dose-dependent manner (Fig. 1B). ADA and CON cells, which showed
lower TrxR1 levels, were more sensitive to auranofin than Mill and
Phi cells (Fig. 1B). Auranofin
completely reduced the level of TrxR1 in ADA cells and this agent
also decreased that of Phi cells (Fig.
1B). In addition, auranofin increased LDH release, sub-G1 cells
and Annexin V positive cells in ADA and Phi cells (Fig. 1C–E). LDH release was high in Phi
cells whereas sub-G1 cells and Annexin V positive cells were high
in ADA cells. It also induced a cleavage in PARP protein in ADA and
Phi cells (Fig. 1F).
Auranofin leads to necrosis in ADA
cells
It was investigated whether auranofin induces
apoptosis and/or necrosis in ADA cells. When auranofin-treated ADA
cells were co-incubated with Z-VAD, a pan-caspase inhibitor, Z-VAD
did not change the percentages of sub-G1 and Annexin V positive
cells in these cells (Fig. 2A and
B). In contrast, NecroX-1, necrosis inhibitor, decreased the
numbers of sub-G1 and Annexin V positive cells in auranofin-treated
ADA cells and necrostatin-1, necroptosis inhibitor, reduced the
numbers of decreased sub-G1 and Annexin V positive cells in these
cells as well (Fig. 2C and D).
NAC prevents auranofin-induced cell death
in ADA and Phi cells
Auranofin is an inhibitor of TrxR1. Therefore, it
can induce cell death through an oxidative stress. We pre-treated
ADA and Phi cells with 2 mM NAC for 1 h prior to the treatment of
auranofin. NAC significantly recovered the reduced cell
proliferation caused by auranofin in ADA and Phi cells (Fig. 3A). NAC also inhibited
auranofin-induced LDH release in both cells (Fig. 3B). NAC significantly prevented cell
death in auranofin-treated ADA and Phi cells, and the prevention
was dramatic in ADA cells (Fig.
3C). As expected, auranofin increased ROS levels including
O2•− in ADA and Phi cells at 24 h, and NAC
decreased the levels in these cells (Fig. 4A and B). Auranofin also induced GSH
depletion in ADA and Phi cells (Fig. 4C
and D). NAC completely blocked the GSH depletion caused by
auranofin in ADA and Phi cells (Fig. 4C
and D).
BSO enhances auranofin-induced cell death
in ADA and Phi cells
There are two main antioxidant systems, Trx and GSH
in cells. For this reason, inhibition of GSH synthesis might be a
novel strategy to disturb the redox status and finally lead to cell
death. As expected, BSO intensified the inhibition of cell
proliferation in Phi cells, which were relatively resistant to
auranofin compared with ADA cells (Fig.
3A). BSO did not additionally increase the LDH release in
auranofin-treated ADA and Phi cells (Fig. 3B). However, BSO significantly
increased apoptotic cell death in auranofin-treated ADA and Phi
cells (Fig. 3C). BSO also augmented
the increased ROS levels including O2•− in
auranofin-treated cells and the augmentation was strong in Phi
cells (Fig. 4A and B). Moreover,
BSO significantly increased the numbers of GSH-depleted cells in
auranofin-treated Phi cells (Fig.
4D).
Discussion
Auranofin as an inhibitor of TrxR has an
anti-inflammatory effect (17) and
can treat rheumatoid arthritis (18). In addition, auranofin shows
anticancer effects in ovarian, prostate, breast and lung cancer
cells (14,19–21).
It is also reported that many cancer cells contain high level of
TrxR expression (6,22). Thus, auranofin can be a strong
candidate agent for treatment of cancer. Likewise, in the present
study, auranofin inhibited the proliferation in mesothelioma cells
and induced caspase-independent apoptosis and necrosis in these
cells. Interestingly, the basal TrxR1 expression levels were not
different between normal mesothelial cells and mesothelioma cells.
This result is contrary to the report that Trx and TrxR were
upregulated in mesothelioma (16).
This discrepancy will be clarified in relation to expression and
activity in Trx and TrxR proteins between normal and cancer cells.
We observed that ADA and CON cells showed low level of TrxR1
expression and these cells were more sensitive to auranofin than
other mesothelioma cells. These results support that the level of
TrxR1 expression is involved in the cytotoxic effectiveness of drug
among cancer cells (23).
Excess ROS production or an imbalance of antioxidant
can lead to oxidative stress and finally damages the cells
(24). Trx and GSH are the two main
antioxidant systems in the cells (25). TrxR is a key component in the Trx
system. Therefore, an inhibition of TrxR can induce cell death via
causing oxidative stress (26).
Correspondingly, auranofin increased the ROS levels including
O2•− in relatively auranofin-sensitive ADA
cells and auranofin-resistant Phi cells. An increase in ROS levels
was strong in Phi cells. This result suggests that
auranofin-resistant Phi cells have a high threshold to oxidative
stress to induce cell death. Furthermore, NAC, an antioxidant,
attenuated the inhibition of proliferation in auranofin-treated ADA
and Phi cells. This agent also prevented cell death in these cells.
The prevention was accompanied by a decrease in ROS levels. These
results suggest that auranofin induce cell growth inhibition and
cell death in an oxidative stress-dependent manner.
GSH is a non-protein antioxidant and prevents cells
from damage caused by oxidative stress (27). The thiol group of cysteine in GSH
supplies an electron to unstable molecules and then GSH itself is
oxidized. When GSH is converted to oxidized-form, it is reduced
back by GSH reductase (28,29). GSH is also critical for cell
proliferation and apoptosis (30,31).
Therefore, an inhibition of GSH is a reasonable strategy to enhance
cytotoxicity in anticancer drug resistant cancer cells (32). Likewise, auranofin increased the
depletion of GSH in both ADA and Phi cells. Auranofin-sensitive ADA
cells showed a strong depletion in GSH content. NAC significantly
blocked GSH depletion in auranofin-treated ADA and Phi cells. Thus,
NAC plays a role as a precursor of GSH as well as an antioxidant in
mesothelioma cells. BSO, an inhibitor of GSH synthesis, intensified
cell growth inhibition and cell death in auranofin-treated ADA and
Phi cells. The enhancement of cell death by BSO was remarkable in
auranofin-resistant Phi cells. BSO also accelerated the increase in
ROS level and GSH depletion in auranofin-treated Phi cells. These
results demonstrated that an inhibition of GSH is effective to
enhance cell growth inhibition and cell death in
auranofin-resistant mesothelioma cells.
In conclusion, it is the first report that auranofin
inhibited cell proliferation in mesothelioma cells and induced cell
death in these cells through an oxidative stress. In addition,
mesothelioma cell death caused by auranofin was affected by the
status of GSH content.
Acknowledgments
We thank Professor Peter R. Hoffmann and Dr Pietro
Bertino for kindly providing the mesothelioma cells. The present
study was supported by the National Research Foundation of Korea
(NRF) grant funded by the Korea government (MSIP) (no.
2008-0062279) and supported by the Basic Science Research Program
through the NRF funded by the Ministry of Education
(2013006279).
Abbreviations:
|
TrxR
|
thioredoxin reductase
|
|
ROS
|
reactive oxygen species
|
|
GSH
|
glutathione
|
|
NADPH
|
nicotinamide adenine dinucleotide
phosphate
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
FBS
|
fetal bovine serum
|
|
MTT
|
3-(4,5-dimethyl thiazol-
2-yl)-2,5-diphenyltetrazolium bromide
|
|
PI
|
propidium iodide
|
|
NAC
|
N-acetylcysteine
|
|
BSO
|
L-buthionine sulfoximine
|
|
LDH
|
lactate dehydrogenase
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
FITC
|
fluorescein isothiocyanate
|
|
GSH
|
glutathione
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
Z-VAD-FMK
|
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
|
References
|
1
|
Lu J and Holmgren A: The thioredoxin
antioxidant system. Free Radic Biol Med. 66:75–87. 2014. View Article : Google Scholar
|
|
2
|
Chen B, Nelin VE, Locy ML, Jin Y and
Tipple TE: Thioredoxin-1 mediates hypoxia-induced pulmonary artery
smooth muscle cell proliferation. Am J Physiol Lung Cell Mol
Physiol. 305:L389–L395. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bobba A, Casalino E, Petragallo VA and
Atlante A: Thioredoxin/thioredoxin reductase system involvement in
cerebellar granule cell apoptosis. Apoptosis. 19:1497–1508. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Laborde E: Glutathione transferases as
mediators of signaling pathways involved in cell proliferation and
cell death. Cell Death Differ. 17:1373–1380. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cadenas C, Franckenstein D, Schmidt M,
Gehrmann M, Hermes M, Geppert B, Schormann W, Maccoux LJ, Schug M,
Schumann A, et al: Role of thioredoxin reductase 1 and thioredoxin
interacting protein in prognosis of breast cancer. Breast Cancer
Res. 12:R442010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iwasawa S, Yamano Y, Takiguchi Y, Tanzawa
H, Tatsumi K and Uzawa K: Upregulation of thioredoxin reductase 1
in human oral squamous cell carcinoma. Oncol Rep. 25:637–644.
2011.PubMed/NCBI
|
|
7
|
Fu JN, Li J, Tan Q, Yin HW, Xiong K, Wang
TY, Ren XY and Zeng HH: Thioredoxin reductase inhibitor ethaselen
increases the drug sensitivity of the colon cancer cell line LoVo
towards cisplatin via regulation of G1 phase and reversal of G2/M
phase arrest. Invest New Drugs. 29:627–636. 2011. View Article : Google Scholar
|
|
8
|
Lin T, Ding Z, Li N, Xu J, Luo G, Liu J
and Shen J: 2-Tellurium-bridged β-cyclodextrin, a thioredoxin
reductase inhibitor, sensitizes human breast cancer cells to
TRAIL-induced apoptosis through DR5 induction and NF-κB
suppression. Carcinogenesis. 32:154–167. 2011. View Article : Google Scholar
|
|
9
|
Liang YW, Zheng J, Li X, Zheng W and Chen
T: Selenadiazole derivatives as potent thioredoxin reductase
inhibitors that enhance the radiosensitivity of cancer cells. Eur J
Med Chem. 84:335–342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kiebala M, Skalska J, Casulo C, Brookes
PS, Peterson DR, Hilchey SP, Dai Y, Grant S, Maggirwar SB and
Bernstein SH: Dual targeting of the thioredoxin and glutathione
antioxidant systems in malignant B cells: A novel synergistic
therapeutic approach. Exp Hematol. 43:89–99. 2015. View Article : Google Scholar
|
|
11
|
Isakov E, Weisman-Shomer P and Benhar M:
Suppression of the pro-inflammatory NLRP3/interleukin-1β pathway in
macrophages by the thioredoxin reductase inhibitor auranofin.
Biochim Biophys Acta. 1840:3153–3161. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu N, Li X, Huang H, Zhao C, Liao S, Yang
C, Liu S, Song W, Lu X, Lan X, et al: Clinically used antirheumatic
agent auranofin is a proteasomal deubiquitinase inhibitor and
inhibits tumor growth. Oncotarget. 5:5453–5471. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park SH, Lee JH, Berek JS and Hu MC:
Auranofin displays anticancer activity against ovarian cancer cells
through FOXO3 activation independent of p53. Int J Oncol.
45:1691–1698. 2014.PubMed/NCBI
|
|
14
|
Fan C, Zheng W, Fu X, Li X, Wong YS and
Chen T: Enhancement of auranofin-induced lung cancer cell apoptosis
by selenocystine, a natural inhibitor of TrxR1 in vitro and in
vivo. Cell Death Dis. 5:e11912014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mansfield AS, Roden AC, Peikert T, Sheinin
YM, Harrington SM, Krco CJ, Dong H and Kwon ED: B7-H1 expression in
malignant pleural mesothelioma is associated with sarcomatoid
histology and poor prognosis. J Thorac Oncol. 9:1036–1040. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kahlos K, Soini Y, Säily M, Koistinen P,
Kakko S, Pääkkö P, Holmgren A and Kinnula VL: Up-regulation of
thioredoxin and thioredoxin reductase in human malignant pleural
mesothelioma. Int J Cancer. 95:198–204. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han S and Kim K, Kim H, Kwon J, Lee YH,
Lee CK, Song Y, Lee SJ, Ha N and Kim K: Auranofin inhibits
overproduction of pro-inflammatory cytokines, cyclooxygenase
expression and PGE2 production in macrophages. Arch Pharm Res.
31:67–74. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suarez-Almazor ME, Spooner CH, Belseck E
and Shea B: Auranofin versus placebo in rheumatoid arthritis.
Cochrane Database Syst Rev. 2:CD0020482000.PubMed/NCBI
|
|
19
|
Kim NH, Park HJ, Oh MK and Kim IS:
Antiproliferative effect of gold(I) compound auranofin through
inhibition of STAT3 and telomerase activity in MDA-MB 231 human
breast cancer cells. BMB Rep. 46:59–64. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park N and Chun YJ: Auranofin promotes
mitochondrial apoptosis by inducing annexin A5 expression and
translocation in human prostate cancer cells. J Toxicol Environ
Health A. 77:1467–1476. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Papaioannou M, Mylonas I, Kast RE and
Brüning A: Disulfiram/copper causes redox-related proteotoxicity
and concomitant heat shock response in ovarian cancer cells that is
augmented by auranofin-mediated thioredoxin inhibition.
Oncoscience. 1:21–29. 2014.
|
|
22
|
Lincoln DT, Al-Yatama F, Mohammed FM,
Al-Banaw AG, Al-Bader M, Burge M, Sinowatz F and Singal PK:
Thioredoxin and thioredoxin reductase expression in thyroid cancer
depends on tumour aggressiveness. Anticancer Res. 30:767–775.
2010.PubMed/NCBI
|
|
23
|
Eriksson SE, Prast-Nielsen S, Flaberg E,
Szekely L and Arnér ES: High levels of thioredoxin reductase 1
modulate drug-specific cytotoxic efficacy. Free Radic Biol Med.
47:1661–1671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ferrer MD, Sureda A, Tauler P, Palacín C,
Tur JA and Pons A: Impaired lymphocyte mitochondrial antioxidant
defences in variegate porphyria are accompanied by more inducible
reactive oxygen species production and DNA damage. Br J Haematol.
149:759–767. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Scarbrough PM, Mapuskar KA, Mattson DM,
Gius D, Watson WH and Spitz DR: Simultaneous inhibition of
glutathione- and thioredoxin-dependent metabolism is necessary to
potentiate 17AAG-induced cancer cell killing via oxidative stress.
Free Radic Biol Med. 52:436–443. 2012. View Article : Google Scholar
|
|
26
|
Lopert P, Day BJ and Patel M: Thioredoxin
reductase deficiency potentiates oxidative stress, mitochondrial
dysfunction and cell death in dopaminergic cells. PLoS One.
7:e506832012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dunning S, Ur Rehman A, Tiebosch MH,
Hannivoort RA, Haijer FW, Woudenberg J, van den Heuvel FA,
Buist-Homan M, Faber KN and Moshage H: Glutathione and antioxidant
enzymes serve complementary roles in protecting activated hepatic
stellate cells against hydrogen peroxide-induced cell death.
Biochim Biophys Acta. 1832:2027–2034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iversen R, Andersen PA, Jensen KS, Winther
JR and Sigurskjold BW: Thiol-disulfide exchange between
glutaredoxin and glutathione. Biochemistry. 49:810–820. 2010.
View Article : Google Scholar
|
|
29
|
Kumar C, Igbaria A, D'Autreaux B, Planson
AG, Junot C, Godat E, Bachhawat AK, Delaunay-Moisan A and Toledano
MB: Glutathione revisited: A vital function in iron metabolism and
ancillary role in thiolredox control. EMBO J. 30:2044–2056. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schoeneberger H, Belz K, Schenk B and
Fulda S: Impairment of antioxidant defense via glutathione
depletion sensitizes acute lymphoblastic leukemia cells for Smac
mimetic-induced cell death. Oncogene. 34:4032–4043. 2015.
View Article : Google Scholar
|
|
31
|
Buşu C, Li W, Caldito G and Aw TY:
Inhibition of glutathione synthesis in brain endothelial cells
lengthens S-phase transit time in the cell cycle: Implications for
proliferation in recovery from oxidative stress and endothelial
cell damage. Redox Biol. 1:131–139. 2013. View Article : Google Scholar
|
|
32
|
Hall MD, Marshall TS, Kwit AD, Miller
Jenkins LM, Dulcey AE, Madigan JP, Pluchino KM, Goldsborough AS,
Brimacombe KR, Griffiths GL, et al: Inhibition of glutathione
peroxidase mediates the collateral sensitivity of
multidrug-resistant cells to tiopronin. J Biol Chem.
289:21473–21489. 2014. View Article : Google Scholar : PubMed/NCBI
|