Introduction
Numerous studies have demonstrated that selenium is
an essential trace element, pivotal for human health (1,2). Serum
selenium levels are correlated with the incidence of many diseases,
of which cancer attracts the utmost attention (3–5).
Numerous preclinical and epidemiological studies have demonstrated
the chemopreventive efficacy of selenium against cancers (6,7).
Sodium selenite, an inorganic form of selenium, has been shown to
induce cancer cell death via various mechanisms (8,9).
Numerous studies including ours, have shown that sodium selenite
induces the apoptosis of malignant cancer cells such as leukemia,
colorectal, lung and prostate cancer (10,11),
yet the detailed mechanisms of how selenite induces cell death are
far from clear. Colorectal cancer (CRC) is the second leading cause
of cancer-related patient death in the US. Thus, there is an urgent
need for novel drugs for CRC (12).
Autophagy is an eukaryotic conserved degradative
system. When cells are confronted with stress, they form
double-membrane autophagosomes to constrain superfluous organelles
or long-lived proteins. The crosstalk between autophagy and
apoptosis is complicated (13,14).
Various studies show that autophagy may cooperate with apoptosis to
induce cell death (15–17). We aimed to investigate the
relationship between autophagy and apoptosis in selenite-treated
CRC cells.
We previously showed that supranutritional sodium
induced apoptosis in leukemia and CRC cells (18–20).
In the present study, we conducted a series of experiments to
unveil the role of autophagy and apoptosis in selenite-treated CRC
cells. We validated that sodium selenite induced protective
autophagy in CRC cells. Inhibition of autophagy enhanced the
apoptosis of the CRC cells, while inhibition of apoptosis resulted
in decreased autophagy. These results showed that sodium selenite
induced both autophagy and apoptosis in the CRC cells. However, the
detailed mechanism underlying the crosstalk between
selenite-induced autophagy and apoptosis in CRC cells warrants
further study.
Materials and methods
Cells and antibodies
HCT116 and SW480 CRC cells were maintained in
Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Paisley,
Scotland, UK) supplemented with 10% fetal bovine serum (FBS)
(HyClone, Logan, UT, USA), and antibiotics (100 U/ml penicillin and
100 µg/ml streptomycin) in a humidified 5% CO2
atmosphere at 37°C. Sodium selenite was purchased from
Sigma-Aldrich (St. Louis, MO, USA). Antibodies against cleaved
caspase-9, cleaved PARP, LC3 or Beclin-1 were purchased from Cell
Signaling Technology (Danvers, MA, USA). Antibodies to β-actin were
purchased from Sigma-Aldrich. The p62 antibody was purchased from
Abcam (Cambridge, MA, USA).
Protein isolation and immunoblot
analysis
Cells were lysed in RIPA buffer (20 mM Tris pH 7.5,
150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium
pyrophosphate, 1 mM β-glycerolphosphate, 1 mM
Na3VO4, 1 µg/ml leupeptin and 1 mM
PMSF). The total cell lysates were sonicated and collected by
centrifugation prior to concentration determination using the
Bradford method. The proteins were resolved on 8–15% SDS-PAGE, and
then electro-transferred to nitrocellulose membranes. Subsequently,
the blots were incubated with the indicated primary antibodies and
the corresponding HRP-conjugated secondary antibodies. The
immunoreactive bands were visualized by chemiluminescent reagents
from Thermal Scientific.
Immunofluorescence
Cells were grown on glass slides for 24 h before
treatment with 10 µM selenite for 24 h. The cells were
incubated with LC3 primary antibodies overnight at 4°C, and were
then incubated with FITC fluorescence-labeled secondary antibodies
for 1 h at room temperature, followed by staining with DAPI
solution to visualize the cell nuclei. The punctate of LC3 protein
before and after treatment with selenite in the CRC cells was
detected by an Olympus laser scanning confocal FV1000 microscope
(Olympus, Tokyo, Japan) and analyzed by Olympus FluoView
software.
Plasmid transfection
GFP-LC3 plasmids were transfected into HCT116 and
SW480 CRC cells using Lipofectamine 2000 according to the
manufacturer's instructions. After another 24 h, the cells were
treated with selenite or phosphate-buffered saline (PBS) as a
solution control. The transfection efficiency was confirmed by
western blotting.
Detection of apoptosis by Annexin
V/propidium iodide (PI) double staining
The apoptotic rates of cells were determined using
an Annexin V/PI double staining kit (Merck, Germany) according to
the manufacturer's instructions. Then the cells were subjected to
analysis by a C6 Accuri flow cytometer.
Transmission electron microscopy
(TEM)
TEM was used to observe autophagy and
ultrastructural changes in the HCT116 and SW480 cells 24 h after
selenite treatment. Fixed cells were post-fixed in 2%
OsO4, dehydrated in graded alcohol and flat-embedded in
Epon 812 (Electron Microscopy Sciences, Fort Washington, PA, USA).
Ultra-thin sections (100 nm) were prepared, stained with uranyl
acetate and lead citrate, and examined under an electron microscope
(H-600; Hitachi, Japan).
Ethics statement
The present study was approved by the Ethics
Committee of the Institute of Basic Medical Science. Principles of
laboratory animal care were followed and complied with standards
equivalent to the guidelines for the welfare of animals in
experimental neoplasia.
Mouse xenograft tumor models
BALB/c nude mice (4 weeks old) were purchased from
the Institute of Laboratory Animal sciences. Twenty-eight nu/nu
mice were randomly assigned to four groups and subcutaneously
injected with HCT116 or SW480 CRC cells which were suspended in
serum-free DMEM at a concentration of 2×107 cells/ml in
the left shoulder of the nude mice. After the tumors were palpable,
half of the mice were injected i.p. with sodium selenite (2
mg/kg/day). The control group was injected with 0.9% sodium
chloride, at a volume of ~200 µl/20 g/day. At the end of the
experiment, the mice were sacrificed by cervical dislocation, and
the tumors and livers were rapidly removed and weighed.
Immunohistochemical staining
Tumor tissues from the control and selenite-treated
groups were sectioned and deparaffinized in xylene and dehydrated
with graded ethanols in accordance with the routine method. The
slides were incubated with primary antibodies against cleaved
caspase-9, Beclin-1, p62 or LC3 overnight at 4°C. After being
washed in PBS, the slides were incubated with HRP-conjugated
secondary antibody at room temperature for 2 h treated with
diaminobenzidine working solution, and then counterstained with
Mayer's hematoxylin for 1 min. Finally, the slides were dehydrated
with increasing concentrations of ethanol and clarified with
xylene.
Statistical analysis
All of the above experiments were repeated at least
three times. The results are expressed as the mean ± SD (n≥3). In
addition, Student's t-test was applied to assess the statistically
significant difference (P<0.05).
Results
Selenite treatment induces autophagy in
CRC cells
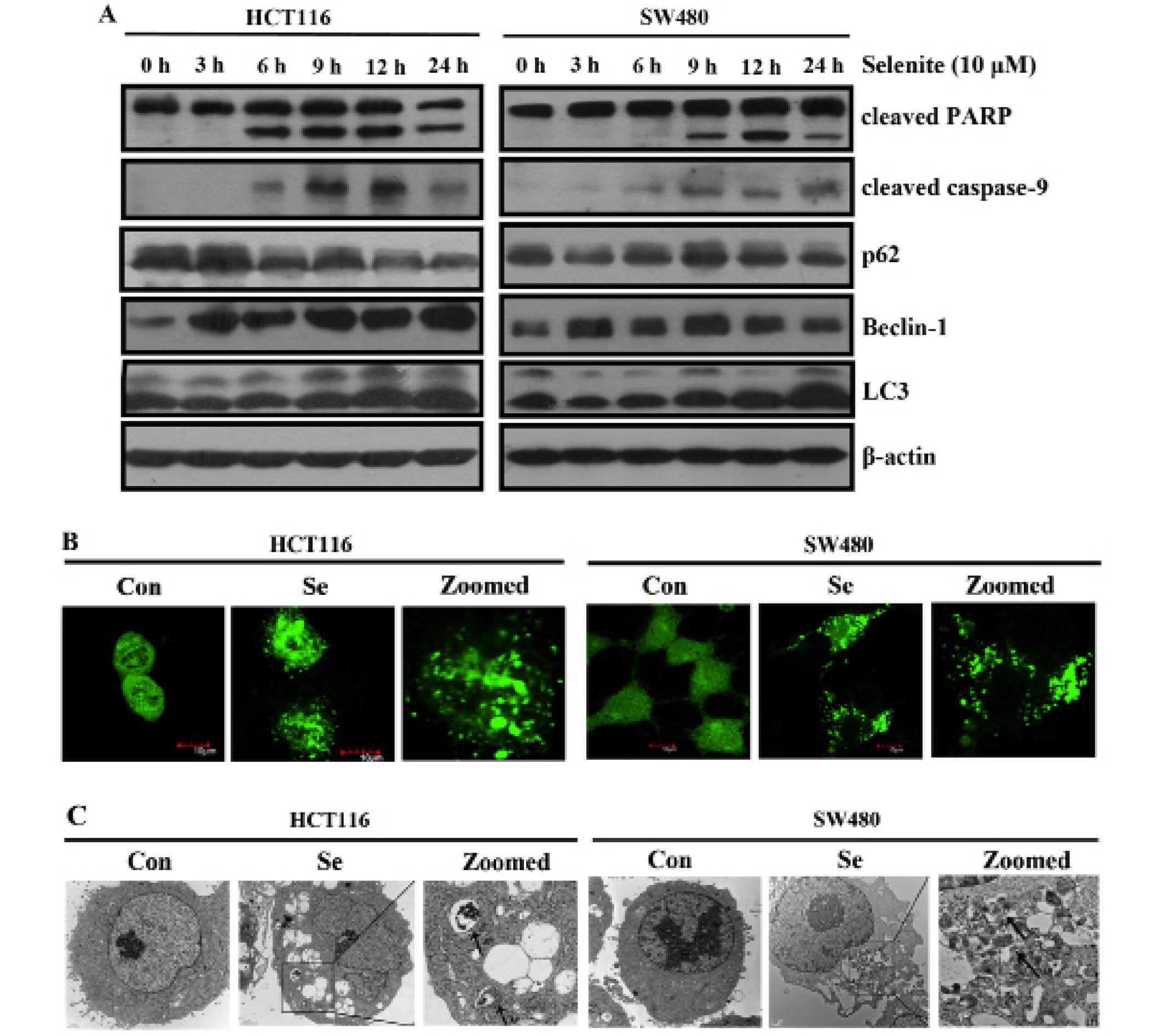
We previously showed that supranutrional selenite
treatment induced apoptosis in HCT116 and SW480 CRC cells (21). As shown in Fig. 1A, the expression of the autophagy
markers and Beclin-1, was increased. p62 is a specific substrate of
autophagy, which was decreased in response to selenite treatment.
We detected the conversion of microtubule-associated protein light
chain 3 (LC3) (from LC3-I to LC3-II) in response to selenite
treatment. Increased punctate of GFP-LC3 was noted in the cells
treated with selenite (Fig. 1B).
Consistently, from the electron micros-copy results (Fig. 1C), we observed more autophagosomes
in the selenite-treated CRC cells. All the results collectively
showed that sodium selenite treatment increased autophagy in the
HCT116 and SW480 CRC cells.
Autophagy acts as a pro-survival
mechanism in the selenite-treated CRC cells
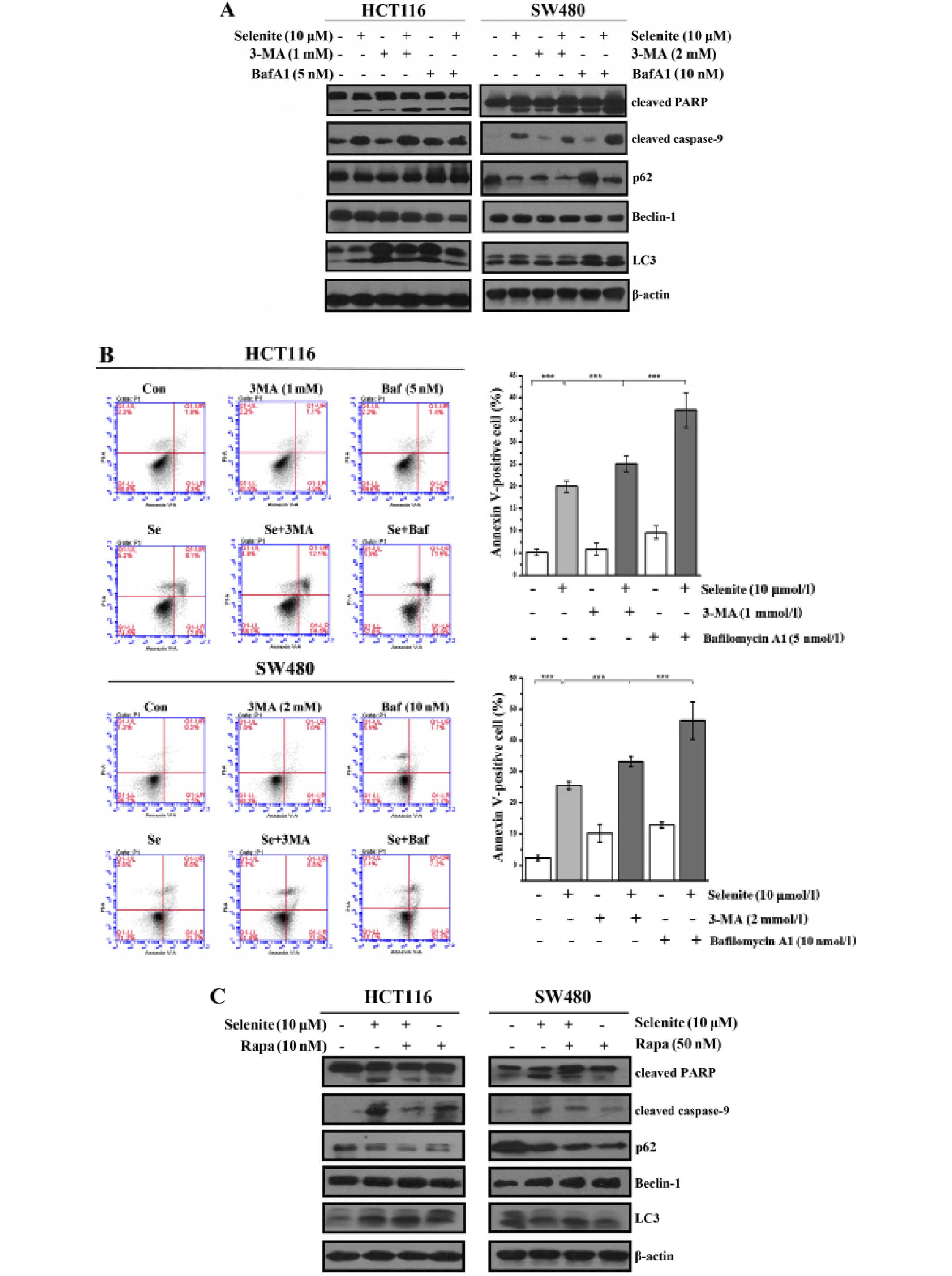
3-Methyladenine, an inhibitor of autophagy
initiation (22), was used to
inhibit selenite-induced autophagy. Bafilomycin A1 was used to
inhibit the degradation of autophagosomes by lysosomes (23). Using western blotting, we showed
that the conversion of LC3 was reduced in the 3-MA-treated samples,
and in contrast, however, LC3-II was accumulated in the bafilomycin
A1-treated cells. From the western blot results, cleaved PARP and
cleaved caspase-9 were greatly increased when autophagy was
inhibited compared with selenite treatment alone (Fig. 2A). Additionally, from the Annexin
V/PI double staining assay (Fig.
2B), we also concluded that when autophagy was inhibited, the
apoptotic rate increased. Rapamycin is widely used to activate
autophagy through its inhibitory effect on mTOR. From the western
blot results, we discovered that LC3 conversion was increased in
the rapamycin-treated cells compared with the control or selenite
treatment group. By detecting cleaved PARP and caspase-9, we found
that the levels of cleaved caspase-9 and PARP were decreased when
autophagy was activated (Fig. 2C).
In accordance with the western blot results using Annexin V/PI
double staining assays, we found that the apoptotic rate was
decreased from 30.1 to 15.2% and 31.1 to 10.3% in the HCT116 and
SW480 cells, respectively (Fig.
2D). When the cells were treated with pancaspase inhibitor,
Z-VAD-fmk, the punctate of LC3 disappeared compared with the
control (Fig. 2E). This
demonstrated that selenite-induced autophagy was elicited by
selenite when apoptosis was induced in CRC cells. These results
indicate that autophagy may facilitate the survival of cells by
antagonizing the proapoptotic effect of selenite.
Sodium selenite treatment inhibits tumor
growth and induces apoptosis and autophagy in HCT116 and SW480
colorectal xenograft models
To further investigate the effect of sodium selenite
on the growth of tumors in xenograft models, we subcutaneously
inoculated HCT116 and SW480 cells in nude mice and developed tumors
to a palpable size. Sodium selenite diet was given daily. After 21
days, the tumors were extracted and photographed. The results are
shown in Fig. 3A and B; 2 mg/kg/day
selenite treatment inhibited both HCT116 and SW480 tumor volume.
The tumor weight was analyzed and the results are show in Fig. 3A and B (upper panel). Tumor weight
in the 2 mg/kg/day selenite treatment group was significantly
decreased compared with the control. Moreover, sodium selenite
treatment had no obvious effect on the body weight of the mice
(Fig. 3C and D). By H&E
staining of the tumor and liver, compared with the control group,
the selenite-treated group showed more pathological changes
including some necrotic and apoptotic cells. Both HCT116 and SW480
tumors from the control group showed uniform large polymorphic,
hyperchromatic spindle-shaped cells and irregularly dispersed
chromatin with a high nuclear/cytoplasmic ratio (Fig. 3E). These results collectively showed
that selenite treatment inhibited tumor growth in both the HCT116
and SW480 colon xenograft models.
To further analyze the effect of selenite on
apoptosis and autophagy in the xenograft models, we exploited
western blotting and immunohistochemical assays to analyze changes
in levels of apoptosis and autophagy markers in the tissues. More
cleaved PARP and caspase-9 were observed in the selenite-treated
samples (Fig. 3G). Autophagy
markers, Beclin-1 and LC3 were increased in the context of selenite
treatment. Consistently, p62 was downregulated in the
selenite-treated tumors, and these results were consistence with
those in the cell culture experiments. Additionally, in the
immunohistochemical experiments (Fig.
3F) we also verified the conclusion that selenite treatment
induced apoptosis and autophagy in the xenograft tumors.
Reactive oxygen species (ROS) play a
pivotal role in selenite-induced apoptosis and autophagy in CRC
cells
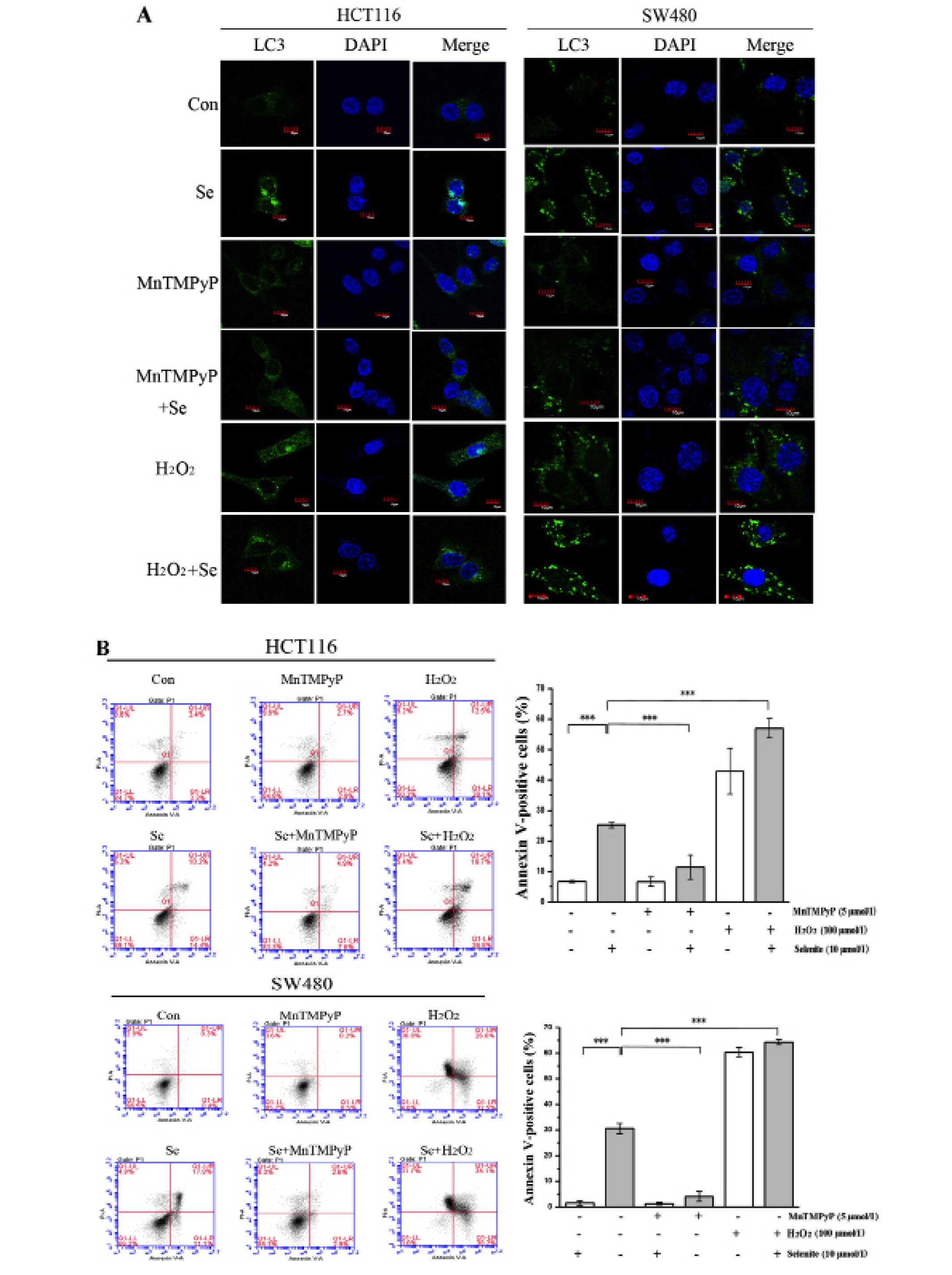
To explore the effect of ROS on selenite-induced
apoptosis and autophagy, we modulated the ROS level in cells using
MnTMPyP and H2O2. When ROS in CRC cells were
scavenged with MnTMPyP, the punctate of LC3 disappeared (Fig. 4A) and cleaved PARP was decreased
significantly even in the presence of selenite treatment (Fig. 4C). While the cells were pretreated
with H2O2 to augment ROS level, we observed
increased punctate of LC3 from confocal (Fig. 4A) and increased PARP cleavage from
western blot results (Fig. 4C).
Accordingly, from Annexin V/PI double staining assay, we found that
the apoptotic rate was decreased when MnTMPyP was added. The
opposite results were shown when using H2O2
compared with the MnTMPyP-treated cells (Fig. 4B). Finally, we detected the change
in the autophagy marker LC3 in the samples treated with MnTMPyP and
found a decrease in the conversion of LC3. We also observed an
opposite trend of change in the H2O2-treated
cells (Fig. 4C). We concluded that
selenite-induced apoptosis and autophagy may be caused by ROS
through some unknown mechanism.
Discussion
In the present study, we found that sodium selenite
induced apoptosis and autophagy in colorectal cancer (CRC) cells.
Investigation into the molecular mechanism underlying the crosstalk
between apoptosis and autophagy bears great significance,
particularly for exploiting novel therapies for treating malignant
cancer. The relationship between apoptosis and autophagy is very
complicated. Autophagy plays an essential role in maintaining cell
survival under stress. In some cases, autophagy acts as a
pro-survival factor via antagonizing apoptosis. Excessive autophagy
also leads to cell death. In our system, we demonstrated that
autophagy was activated by selenite treatment. Suppression of
autophagy in CRC cells augmented the apoptotic rate in the cells.
In the context of further autophagy activation, the apoptotic rate
was increased compared with the selenite treatment (Fig. 4D).
Autophagy has been implicated in a plethora of
physiological and pathological processes (24,25).
It is commonly thought to be activated in cancer cells to sustain
carcinogenesis. However, studies also show that autophagy leads to
cell death if it persists (26,27).
Our previous study showed that sodium selenite induced apoptotic
cell death in CRC cells (21). In
the present study, we discovered that autophagy was activated. This
prompted us to investigate the role of autophagy in
selenite-treated CRC cells. We examined the apoptotic rate of CRC
cells when selenite-induced autophagy was inhibited by the
inhibitor 3-MA and bafilomycin A1. Accordingly, when autophagy was
enhanced, cell death decreased. These results support the
conclusion that autophagy was a pro-survival mechanism in the
selenite-treated CRC cells. Moreover, we inhibited the
selenite-induced apoptosis and discovered that it exerted little
effect on autophagy. We concluded that selenite-induced autophagy
was a self-rescue mechanism together with occurrence of apoptosis
when cells were treated with selenite.
Reactive oxygen species (ROS) are considered an
important anticancer factor of many chemotherapy drugs (28–30).
Our previous studies also showed that selenite treatment could
increase ROS in cancer cells (21,31).
Thus, we examined the role of ROS in the crosstalk between
apoptosis and autophagy. On one hand, when ROS were depleted by ROS
scavenger, both apoptosis and autophagy were inhibited; on the
other hand, in the context of H2O2 treatment,
increased ROS caused more apoptosis and autophagy in CRC cells. It
is consistent with other studies that ROS are a pivotal factor in
causing cell death. These results further show that ROS have an
important role in the crosstalk between autophagy and
apoptosis.
Besides the in vitro results, we corroborated
our findings in xenograft models. We found that sodium selenite
treatment inhibited tumor growth in both models.
Immunohistochemical staining of apoptotic and autophagy markers
indicated that sodium selenite increased autophagy and apoptosis in
tumor tissues. Examination of apoptosis and autophagy related
proteins confirmed this conclusion. These results collectively
revealed that sodium selenite induced apoptosis and autophagy both
in vitro and in vivo.
Although we discovered the phenomenon that sodium
selenite induces apoptosis and autophagy in CRC cells, the detailed
molecular mechanism underlying the crosstalk between apoptosis and
autophagy warrants further study. Furthermore, the role of ROS and
the signaling pathway modulating this complicated relationship
between apoptosis and autophagy in selenite-treated CRC cells
require further elucidation. This research may undoubtedly help to
elucidate the anticancer mechanisms of selenium. These results
provide a theoretical background for the clinical application of
selenium.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (nos. 31170788, 31340037 and
31271565), the National Natural Science Foundation for Young
Scholars of China (no. 31101018), the State Key Laboratory Special
Fund (no. 2060204), and the Natural Science Foundation of Beijing
(no. 5082015).
References
|
1
|
Mistry HD, Broughton Pipkin F, Redman CW
and Poston L: Selenium in reproductive health. Am J Obstet Gynecol.
206:21–30. 2012. View Article : Google Scholar
|
|
2
|
Rayman MP: Selenium and human health.
Lancet. 379:1256–1268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hamdy SM, Latif AK, Drees EA and Soliman
SM: Prevention of rat breast cancer by genistin and selenium.
Toxicol Ind Health. 28:746–757. 2012. View Article : Google Scholar
|
|
4
|
Ou Y, Jiang B, Wang X, Ma W and Guo J:
Selenium and colorectal adenomas risk: A meta-analysis. Nutr
Cancer. 64:1153–1159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hurst R, Hooper L, Norat T, Lau R, Aune D,
Greenwood DC, Vieira R, Collings R, Harvey LJ, Sterne JA, et al:
Selenium and prostate cancer: Systematic review and meta-analysis.
Am J Clin Nutr. 96:111–122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lotan Y, Goodman PJ, Youssef RF, Svatek
RS, Shariat SF, Tangen CM, Thompson IM Jr and Klein EA: Evaluation
of vitamin E and selenium supplementation for the prevention of
bladder cancer in SWOG coordinated SELECT. J Urol. 187:2005–2010.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Klein EA, Thompson IM Jr, Tangen CM,
Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL,
Gaziano JM, et al: Vitamin E and the risk of prostate cancer: The
Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA.
306:1549–1556. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Králová V, Benešová S, Cervinka M and
Rudolf E: Selenite-induced apoptosis and autophagy in colon cancer
cells. Toxicol In Vitro. 26:258–268. 2012. View Article : Google Scholar
|
|
9
|
Guo F, Monsefi N, Moritz A and
Beiras-Fernandez A: Selenium and cardiovascular surgery: An
overview. Curr Drug Saf. 7:321–327. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang C, Hu H, Malewicz B, Wang Z and Lü
J: Selenite-induced p53 Ser-15 phosphorylation and caspase-mediated
apoptosis in LNCaP human prostate cancer cells. Mol Cancer Ther.
3:877–884. 2004.PubMed/NCBI
|
|
11
|
Sanmartín C, Plano D, Sharma AK and Palop
JA: Selenium compounds, apoptosis and other types of cell death: An
overview for cancer therapy. Int J Mol Sci. 13:9649–9672. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kitisin K and Mishra L: Molecular biology
of colorectal cancer: New targets. Semin Oncol. 33(Suppl 11):
S14–S23. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rubinstein AD and Kimchi A: Life in the
balance - a mechanistic view of the crosstalk between autophagy and
apoptosis. J Cell Sci. 125:5259–5268. 2012. View Article : Google Scholar
|
|
15
|
Booth LA, Tavallai S, Hamed HA,
Cruickshanks N and Dent P: The role of cell signalling in the
crosstalk between autophagy and apoptosis. Cell Signal. 26:549–555.
2014. View Article : Google Scholar :
|
|
16
|
Su M, Mei Y and Sinha S: Role of the
crosstalk between autophagy and apoptosis in cancer. J Oncol.
2013(102735)2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fimia GM and Piacentini M: Regulation of
autophagy in mammals and its interplay with apoptosis. Cell Mol
Life Sci. 67:1581–1588. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luo H, Yang Y, Duan J, Wu P, Jiang Q and
Xu C: PTEN-regulated AKT/FoxO3a/Bim signaling contributes to
reactive oxygen species-mediated apoptosis in selenite-treated
colorectal cancer cells. Cell Death Dis. 4:e4812013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ren Y, Huang F, Liu Y, Yang Y, Jiang Q and
Xu C: Autophagy inhibition through PI3K/Akt increases apoptosis by
sodium selenite in NB4 cells. BMB Rep. 42:599–604. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li J, Zuo L, Shen T, Xu CM and Zhang ZN:
Induction of apoptosis by sodium selenite in human acute
promyelocytic leukemia NB4 cells: Involvement of oxidative stress
and mitochondria. J Trace Elem Med Biol. 17:19–26. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luo H, Yang Y, Huang F, Li F, Jiang Q, Shi
K and Xu C: Selenite induces apoptosis in colorectal cancer cells
via AKT-mediated inhibition of β-catenin survival axis. Cancer
Lett. 315:78–85. 2012. View Article : Google Scholar
|
|
22
|
Jiang Q, Wang Y, Li T, Shi K, Li Z, Ma Y,
Li F, Luo H, Yang Y and Xu C: Heat shock protein 90-mediated
inactivation of nuclear factor-κB switches autophagy to apoptosis
through becn1 transcriptional inhibition in selenite-induced NB4
cells. Mol Biol Cell. 22:1167–1180. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamamoto A, Tagawa Y, Yoshimori T,
Moriyama Y, Masaki R and Tashiro Y: Bafilomycin A1 prevents
maturation of autophagic vacuoles by inhibiting fusion between
autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E
cells. Cell Struct Funct. 23:33–42. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eskelinen E-L and Saftig P: Autophagy: A
lysosomal degradation pathway with a central role in health and
disease. Biochim Biophys Acta. 1793:664–673. 2009. View Article : Google Scholar
|
|
25
|
Martinet W, Agostinis P, Vanhoecke B,
Dewaele M and De Meyer GR: Autophagy in disease: A double-edged
sword with therapeutic potential. Clin Sci. 116:697–712. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Buchser WJ, Laskow TC, Pavlik PJ, Lin HM
and Lotze MT: Cell-mediated autophagy promotes cancer cell
survival. Cancer Res. 72:2970–2979. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pilarczyk B, Jankowiak D, Tomza-Marciniak
A, Pilarczyk R, Sablik P, Drozd R, Tylkowska A and Skólmowska M:
Selenium concentration and glutathione peroxidase (GSH-Px) activity
in serum of cows at different stages of lactation. Biol Trace Elem
Res. 147:91–96. 2012. View Article : Google Scholar
|
|
29
|
Tinggi U: Selenium: Its role as
antioxidant in human health. Environ Health Prev Med. 13:102–108.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zou YF, Niu PY, Gong ZY and Yuan J: Role
of reactive oxygen species in sodium selenite induced DNA damage in
HepG2 cells. Wei Sheng Yan Jiu. 35:291–293. 2006.In Chinese.
PubMed/NCBI
|
|
31
|
Li ZS, Shi KJ, Guan LY, Cao TM, Jiang Q,
Yang Y and Xu CM: ROS leads to MnSOD upregulation through ERK2
translocation and p53 activation in selenite-induced apoptosis of
NB4 cells. FEBS Lett. 584:2291–2297. 2010. View Article : Google Scholar : PubMed/NCBI
|