Introduction
Endoplasmic reticulum (ER) is an essential cellular
compartment where new secretory proteins are folded and assembled
to their maturation. When suffering various conditions such as
nutrient deprivation, hypoxia, alterations in glycosylation status
and disturbances of calcium flux, the steady state of the ER
environment is disturbed and misfolded or unfolded proteins are
accumulated and aggregated in ER lumen, failure of ER coping with
the excessive proteins load leads to ER stress (1). To reduce damage from ER stress, cells
trigger unfolded protein response (UPR), which signals transient
attenuation of protein translation, degradation of unfolded and
misfolded proteins and the induction of molecular chaperones
(2). The upregulation of molecular
chaperones in response to expression of dominant negative ubiquitin
may contribute to degradation of abnormal proteins (3).
Three known transmembrane sensors of ER stress are
double-stranded RNA-activated protein kinase-like ER kinase (PERK),
inositol-requiring enzyme 1 (IRE1) and activating transcription
factor 6 (ATF6) (4). Under normal
conditions, the luminal domains of these sensors are occupied by
the ER chaperon glucose-regulated protein 78 (GRP78) (5). Upon ER stress, unfolded or misfolded
proteins compete with these sensors for binding GRP78 segregating
GRP78 from those of luminal domains. As a result, these sensors are
activated by inducing phosphorylation and homodimerization of IRE1
and PERK, and relocalization of ATF6 to the Golgi (6,7).
GRP78, is also known as immunoglobulin heavy chain
binding protein (BIP), is a central regulator of ER function, an ER
molecular chaperon, facilitating new protein folding and
assembling, targeting misfolded proteins for degradation, binding
ER Ca2+ and controlling the activation of trans-membrane
ER stress sensors (8,9). Perhaps due to nutrient deprivation and
hypoxia, increasing evidence shows that the UPR is indispensable in
various solid tumors, investigators have reported elevated
expression of UPR targets, such as GRP78 and GRP94 (10,11).
The expression of these genes and other components of UPR are also
correlated with increased malignancy. It seems that some cancer
cells may have adapted to ER stress by activation of UPR without
resulting in apoptosis (12).
The UPR is a cytoprotective response to ER stress,
but excessive or prolonged UPR results in apoptotic cell death.
Many caspase family members participate in the process of ER
stress-induced apoptosis, such as caspase-2, -3, -4, -7, -8 -9 and
-12 (13–17). Among them, caspase-12 is thought to
be a key mediator in rodents. However, caspase-4 has been shown to
play an important role in ER stress-induced apoptosis of human
neuroblastoma and HeLa cells. Mutual action between caspase-4 and
GRP78 suggests that caspase-4 lies in ER lumen (18,19),
this provides structural basis for regulation of ER stress-induced
apoptosis by caspase-4.
Previous studies have shown that survival signaling
pathways, such as the phosphatidylinositol 3-kinase (PI3K)/AKT and
mitogen-activated protein kinase (MAPK)/extracellular signal
regulated kinase (ERK) and (MEK)/ERK pathways, may also play roles
in counteracting the apoptosis-inducing potential of ER stress.
Particularly, constitutive activation of the MEK/ERK pathway is
reported as a common cause for resistance of melanoma cells to
apoptosis mediated by the death receptor and mitochondrial
apoptotic pathways (20–23).
In the present study, we found that UPR was
constitutively activated in the breast cancer cells compared to
normal breast epithelial cells in which GRP78 expression of cancer
cells was obviously higher than epithelial cells. Moreover, we
found that tunicamycin (TM), an ER stress inducer, not only
strongly activated UPR, but also the MEK/ERK pathway, and
inhibition of the pathway sensitized breast cancer cells to
TM-induced apoptotic cell death due to downregulation of GRP78
expression. This sensitization of breast cancer cells to TM-induced
apoptosis by inhibition of MEK/ERK and GRP78 was caspase-dependent,
at least in part, by activation of caspase-4. These results
indicated that GRP78 is a possible chemotherapeutical target and
have important implications for new treatment strategies in breast
cancer by combination of agents that induce ER stress with
inhibitors of the MEK/ERK pathway.
Materials and methods
Cell lines
Human breast cancer cell lines MCF-7, MDA-MB-231 and
SK-BR-3 were cultured in Department of Biochemistry and Molecular
Biology, Nanjing Medical University. Human mammary epithelial cell
line MCF-10A and human pneumonic epithelial cell WI-38 was
purchased from the cell bank of Academia Sinica. They were cultured
at 37°C in the presence of 5% CO2 in Dulbecco's Modified
Eagle's medium (DMEM; Gibco) supplemented with 10% fetal calf serum
(FCS) and 100 U/ml penicillin G and 100 mg/ml streptomycin.
Clinical specimens
Clinical specimens were collected from the patients
registered at the First Affiliated Hospital of Bengbu Medical
College (Bengbu, China) between 2005 and 2008 with the patients
consent and Ethics Committee approval. To monitor GRP78 protein
immunostaining with clinicopathological stages, patient medical
records were retrospectively reviewed at the time when the study
sample was obtained. All samples were randomly selected and
arranged as three groups: breast cancers, adjacent non-cancerous
tissue and breast fibroadenoma.
Antibodies, recombinant proteins and
other reagents
The Vectastain ABC kit and the DAB kit were from
Wuhan Boster Biotechnology Co. Tunicamycin (TM) was purchased from
Sigma Chemical Co. (Castle Hill, Australia), and dissolved in
dimethyl sulfoxide (DMSO) and made to stock solutions of 1 mmol/l.
The rabbit polyclonal antibody against GRP78, ERK1 and ERK2 were
purchased from Santa Cruz Biotechnology Co. The mouse monoclonal
antibody against P-ERK1/2 and β-actin were purchased from Santa
Cruz Biotechnology Co. The mouse IgG and rabbit IgG were purchased
from Sigma Chemical Co. Propidium iodide (PI) was purchased from
Sigma Chemical Co. Reverse transcription-qPCR assay kit was from
Takara Co. TRIzol was from Invitrogen Co. The siGENOME SMART pool
reagents, the siGENOME SMART pool GRP78 (M-008198-01) and control
non-targeting siRNA pool (D-001206-13-20) were obtained from
Dharmacon. Opti-MEM medium and Lipofectamine 2000 reagent were
purchased from Invitrogen Co.
Immunohistochemical staining
Five micrometer-thick sections were cut from the
formalin-fixed, paraffin-embedded block of each case. Sections were
deparaffinized in xylene and rehydrated through graded decreasing
concentrations of alcohol. Antigen was repaired in 0.01 mol/l of
citrate buffer (pH 6.0) by heating in a microwave oven for 5 min
and repeating three times. Rabbit anti-human GRP78 antibody was
added at a dilution of 1:100 in phosphate-buffered saline (PBS) for
1 h at 37°C. The Vectastain ABC kit was used to bind the antibodies
according to the manufacturer's instructions and the binding sites
were visualized using the DAB kit. The sections were counterstained
with Harris hematoxylin. Negative controls were performed by
omission of the primary antibody replaced by PBS in each experiment
and each heavy pigmented case.
Each section was observed in 10 HP visual fields
(VFs), and 200 cells/VF were counted. Score of positive cells
<10% was regarded as 0, 10–50% as 1, 51–75% as 2, and ≥75% as 3.
No staining was 0, weakly positive staining was 1, moderately
positive staining was 2 and strongly positive staining was 3. An
immunoreactive score (IRS) was derived by multiplying the score of
percentage of positive cells with the score of staining intensity.
IRS=0 was regarded as negative; IRS=4 was weakly positive; and IRS
≥4 was strongly positive.
PI uptake assay and apoptosis
Cells were seeded at 1×105/well into
24-well plates and allowed to reach exponential growth for 16–24 h
before treatment. PI staining procedure followed the manufacturer's
protocol. PI buffer was mixed with 100 ml dH2O, 0.005 g
PI, 0.1 g trisodium citrate, 100 µl Triton X-100, covering
with foil and keeping at 4°C. Quantitation of apoptotic cells were
represented by measurement of sub-G1 DNA content using PI staining
in FCM.
Protein extraction and western blot
analysis
Whole cell extracts were obtained by lysing cells in
a Triton X-100-based lysis buffer (10 mmol/l Tris-HCl pH 7.4, 140
mol/l NaCl, 0.5 mmol/l CaCl2, 10.5 mmol/l
MgCl2, 3 mmol/l NaN3, Triton X-100 2 ml,
adding to 12 µmol/l leupeptin, 1 mmol/l PMSF, respectively,
before use. Extraction for phosphorylated proteins needs adding to
50 mmol/l NaF and 1 mmol/l Na3VO4). The
protein content of cell extracts was determined by the BCA assay. A
total load of 30–40 µg proteins was electrophoresed on 10%
SDS-PAGE gels and transferred to PVDF membranes. Membranes were
blocked, incubated with primary antibody at the appropriate
concentration (1:500-1,000), and subsequently incubated with
horseradish peroxidase-conjugated goat anti-rabbit IgG or goat
anti-mouse IgG (1:5,000-10,000 dilution). Labeled bands were
detected by Renaissance Western Blot Chemiluminescence Reagent
(Pierce Co.) and exposed on Hyper MP autoradiography film
(Amersham). The densities of the bands were quantitated by Gel
Imaging System as proteins relatively expression levels.
Reverse transcription and quantitative
PCR (RT-qPCR)
Total RNA was extracted according to the protocol
and RNA concentrations were determined spectrophotometrically at
260 nm. Total RNA (1 µg) was reverse transcribed to cDNA in
a total volume of 20 µl system using a RT reaction kit
(Takara). Quantitative PCR was performed using an Mx3000P Real-Time
PCR system (Applied Biosystems) according to the manufacturer's
instructions and SYBR Premix Ex Taq (Takara) as a DNA-specific
fluorescent dye. PCR was carried out for 40 cycles of 95°C for 10
sec and 60°C for 1 min. All the reactions were repeated three
times. Gene expression levels were calculated relative to GAPDH as
a 2−ΔΔCt value using Stratagene Mx3000P software. Ratio
of the treatment group 2−ΔΔCt value and the negative
group represents relative expression levels. The primers used for
amplification were as follows: GRP78,
5′-GTTTGCTGAGGAAGACAAAAAGCTC-3′ and
5′-CACTTCCATAGAGTTTGCTGATAATTG-3′; GAPDH,
5′-GGGAAGGTGAAGGTCGGAGTC-3′ and 5′-AGCAGAG GGGGCAGAGATGAT-3′.
Primers and probes for GRP78 and GAPDH were from Sangon Co.,
Shanghai, China.
Small RNA interference (RNAi)
transfection
Breast cancer cells MCF-7 and MDA-MB-231 were seeded
at 5×104 cells/well in 24-well plates and allowed to
reach ~50% confluence on the day of transfection. Cells were
transfected with 50–100 nmol/l siRNA in Opti-MEM medium with 10%
FCS using Lipofectamine 2000 reagent according to the
manufacturer's transfection protocol. Twenty-four hours after
transfection, the cells were switched into medium containing 10%
FCS and treated as designed before quantitation of apoptotic cells
by measurement of sub-G1 DNA content using the PI method in FCM.
Efficiency of siRNA transfection was measured by western blot
analysis 24 h later.
Caspase-4 activity assay
Caspase-4 can catalyze substrate acetyl-Leu-Val-Asp
p-nitroanilide (Ac-LEVD-pNA) to flavous
p-nitroaniline (p-NA) with strong absorbance at the
wave-length of 405 nm, thus, absorbance was measured to determine
caspase-4 activity. Total proteins were extracted as previously
described in western blot analysis and quantitated by Bradford
method. Measurement of caspase-4 activity was carried out at the
wavelength of 405 nm, according to the manufacturer's protocol.
Statistical analysis
The statistical software SPSS, version 16.0, and the
Student's t-test were used for statistical analysis. A P-value of
<0.05 was considered to indicate a statistical significance.
Results
GRP78 expression is increased in breast
cancer
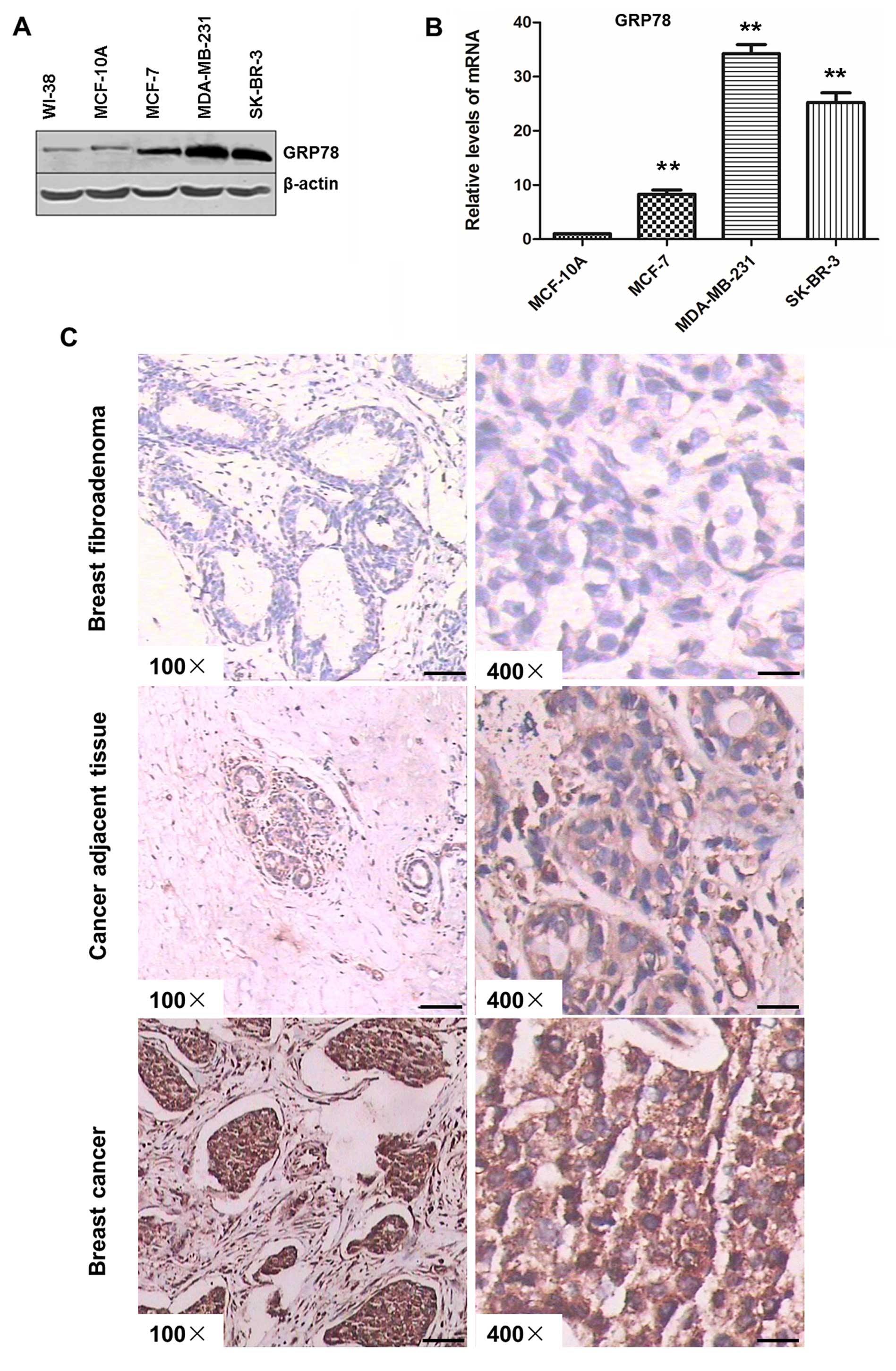
In cell lines, GRP78 expression of breast cancer was
significantly higher than that of normal epidermic cells both in
protein and mRNA levels (Fig. 1A and
B). GRP78 expression in paraffin blocks of breast cancer
tissues using immunohistochemical staining is shown in Fig. 1C, the tissues were from 50 cases of
breast cancer, 50 cases of adjacent non-cancerous tissue and 20
cases of breast fibroadenoma. GRP78 was expressed at significantly
higher levels in breast cancer tissues in comparison with that in
adjacent non-cancerous tissue and breast fibroadenoma (P<0.01,
Dunnette t-test), GRP78 was strongly positive in breast cancer
(84%), weakly positive in adjacent non-cancerous tissue (70%), and
markedly negative in breast fibroadenoma (80%) (Table I). These results indicated that UPR
was constitutively activated in breast cancer.
 | Table IExpression of GRP78 in tissue by
immunohistochemistry. |
Table I
Expression of GRP78 in tissue by
immunohistochemistry.
| Specimen | Cases | GRP78 (IRS)
|
|---|
| 0 (%) | 1–4 (%) | ≥4 (%) |
|---|
| Breast
fibroadenoma | 20 | 16 (80) | 4 (20) | 0 |
| Cancer adjacent
tissue | 50 | 10 (20) | 35 (70) | 5 (10) |
| Breast cancer
tissue | 50 | 1 (2) | 7 (14) | 42 (84) |
Breast cancer cells are relatively
resistant to ER stress-induced apoptosis
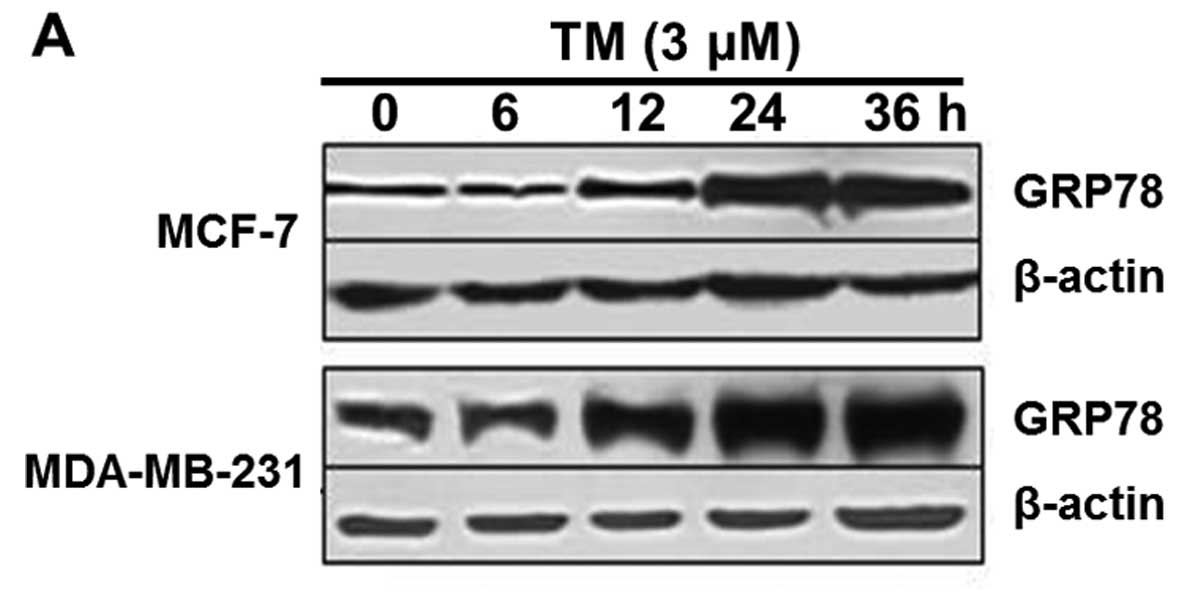
We treated MCF-7, MDA-MB-231 cells with TM, an ER
stress inducer. TM markedly upregulated GRP78 expression and in 24
h the upregulation peaked (Fig.
2A), indicative of further activation of the UPR. At the same
time, we treated MCF-7, MDA-MB-231, MCF-10A and WI-38 cells with
TM, respectively, at a range of concentrations for 48 h to study
the apoptosis induction potential of ER stress. However, the two
breast cancer cell lines were induced from minimal to moderate
levels of apoptosis (<30% apoptotic cells), even when used at
relatively high concentration (TM at 12 µmol/l), which
efficiently killed MCF-10A and WI-38 by induction of apoptosis
(Fig. 2B).
Inhibition of MEK/ERK sensitizes breast
cancer cells to ER stress-induced apoptosis
Constitutive activation of the MEK/ERK pathway is
reported as a commonly acceptable reason for resistance of cancer
cells to apoptosis. Therefore, we monitored if ER stress induced
activation of the MEK/ERK pathway by examining phosphorylation
(activation) of ERK1/2 in whole cell lysates from MCF-7 and
MDA-MB-231 cells with exposure to TM. As shown in Fig. 3A, ERK1/2 was weakly activated in
control cell lines, but the levels of activation were
constitutively increased by treatment with TM, indicating that ER
stress induced further ERK1/2 activation in breast cancer cells. We
next measured the effect of MEK inhibition on TM-induced apoptosis
in breast cancer cells by treating MCF-7 and MDA-MB-231 cells with
the MEK inhibitor U0126 1 h before TM treatment for another 24 h.
As shown in Fig. 3B and C, not only
U0126 inhibited activation of ERK1/2 in the presence or absence of
TM, but it significantly sensitized the cells to TM-induced
apoptosis (P<0.01, Student's t-test). However, the MEK inhibitor
alone did not cause appreciable apoptotic cell death in the cell
lines.
U0126 downregulates GRP78 expression in
both protein and mRNA levels
GRP78 is believed to play an essential role in
protection of cells from ER stress-induced apoptosis (24). Therefore, we deduced that
sensitization of breast cancer cells to ER stress-induced apoptosis
by inhibition of MEK/ERK probably related to regulation of GRP78.
As shown in Fig. 4A exposure to
U0126, a special MEK inhibitor, resulted in reduction in the levels
of GRP78 expression and attenuated its induction by TM in both
MCF-7 and MDA-MB-231 cells. To examine if inhibition of MEK/ERK
blocked transcription of GRP78, we monitored mRNA levels of GRP78
by RT-qPCR in MCF-7 and MDA-MB-231 cells treated with U0126 1 h
before TM treatment. As shown in Fig.
4B, GRP78 mRNA levels in cells treated with TM in the presence
of U0126 were markedly lower than those in cells treated with TM
alone (P<0.01, Student's t-test).
GRP78 knockdown by siRNA sensitizes
breast cancer cells to ER stress-induced apoptosis
To further study if sensitization of breast cancer
cells to ER stress-induced apoptosis was due to reduced GRP78
expression, we used siRNA knock-down of GRP78, then observed ER
stress-induced apoptosis by TM in MCF-7 and MDA-MB-231 cells. As
shown in Fig. 5A exposure to GRP78
siRNA resulted in blockage of GRP78 expression in both MCF-7 and
MDA-MB-231 cells, while control siRNA had no effect on the levels
of GRP78 expression, compared to the negative group. As shown in
Fig. 5B, inhibition of GRP78 by
siRNA resulted in substantial increases in sensitivity of breast
cancer cells to apoptosis induced by TM in the absence of U0126
(P<0.01, Student's t-test). MCF-7 and MDA-MB-231 cells were
treated with U0126 1 h before TM treatment, along with GRP78
knockdown by siRNA, which resulted in more significant increases in
sensitivity of cells to apoptosis (P<0.01, Student's t-test).
However, inhibition of GRP78 by siRNA only failed to induce
significant apoptosis in both MCF-7 and MDA-MB-231 cells.
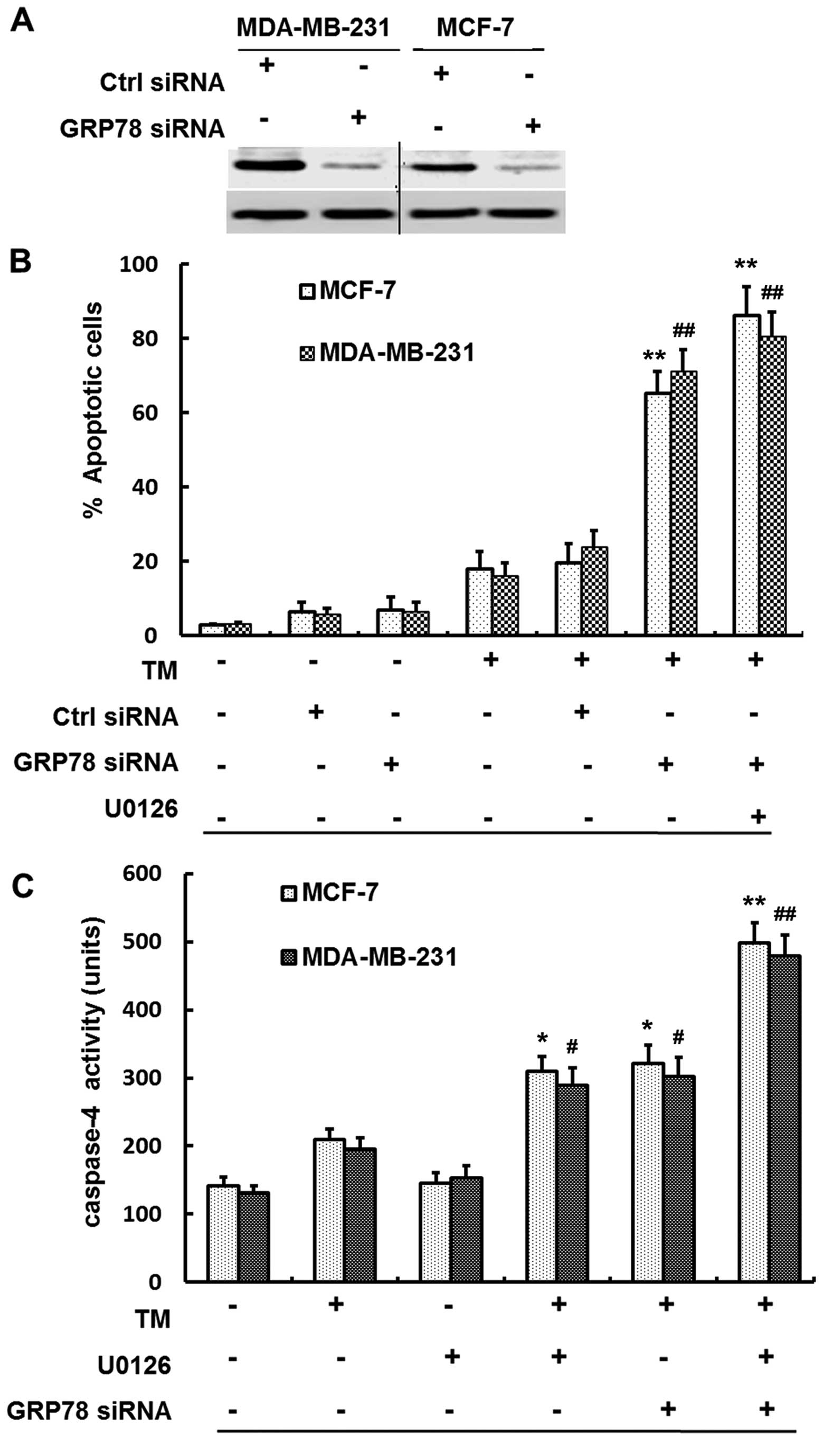 | Figure 5GRP78 knockdown by siRNA sensitizes
breast cancer cells to TM-induced apoptosis and the sensitization
is at least in part caspase-4 dependent. (A) Efficiency of
knockdown of GRP78 by siRNA. Whole-cell lysates from MCF-7 and
MDA-MB-231 cells with control siRNA or GRP78 siRNA transfection for
24 h were subjected to western blot analysis of GRP78 expression.
(B) siRNA knock-down of GRP78 expression sensitized breast cancer
cells to ER stress-induced apoptosis. MCF-7 and MDA-MB-231 cells
were transfected with the control or GRP78 siRNA, 24 h later, the
cells were treated with TM (3 µmol/l) for 48 h or U0126 for
1 h before the addition of TM for further 48 h. Apoptosis was
measured by the PI method using FCM. **P<0.01 for the
two and three combinations of TM, GRP78 siRNA and U0126 compared to
TM, GRP78 siRNA and U0126 alone, respectively, in MCF-7 cells.
##P<0.01 for the same compare in MDA-MB-231 cells.
(C) Sensitization of breast cancer cells to TM-induced apoptosis
was caspase-4 dependent, at least in part. Whole-cell lysates from
MCF-7 and MDA-MB-231 cells treated with U0126 (20 µmol/l),
TM (3 µmol/l), GRP78 siRNA and their combinations of two or
three for 24 h were subjected to caspase-4 activity assay.
*P<0.01 for the combination of TM, GRP78 siRNA or
U0126 compared to TM, GRP78 siRNA and U0126 alone, respectively, in
MCF-7 cells; #P<0.01 for the same compared with
MDA-MB-231 cells. **P<0.01 for the three combination
of TM, GRP78 siRNA and U0126 com-pared to TM, GRP78 siRNA and U0126
alone, respectively, in MCF-7 cells. ##P<0.01 for the
same compared with MDA-MB-231 cells. |
Caspase-4 activity was monitored to examine whether
sensitization of breast cancer cells to TM-induced apoptosis by
inhibition of MEK/ERK is caspase-dependent. As shown in Fig. 5C, tunicamycin (TM) or U0126 alone
did not constitutively activate caspase-4, whereas, TM along with
U0126 markedly activated it in that caspase-4 activity was
increased by 2-fold (P<0.05, Student's t-test). Furthermore,
knockdown of GRP78 by siRNA and TM caused an increase of 2-fold
caspase-4 activity (P<0.05, Student's t-test). Altogether GRP78
siRNA with TM in the presence of U0126, caspase-4 was 4-fold
increased in both MCF-7 and MDA-MB-231 cells (P<0.01, Student's
t-test).
Discussion
Resistance to chemotherapy is a major obstacle to
improving therapeutic effects of breast cancer. The study of
resistance mechanisms is becoming a new strategy of overcoming
various cancers. The above results suggest that there are two
apoptotic resistance mechanisms in breast cancer which are
constitutively activation of MEK/ERK pathway and stress induction
of the UPR. GRP78 was found to be expressed at relatively high
levels in cultured breast cancer cells and tissue, but were at low
levels in cultured breast normal epithelial cells and normal
tissue, which suggest that the UPR may have tumor-specific
selectivity. The strongly positive GRP78 expression suggests that
the UPR is acutely activated in breast cancer and targeted therapy
against breast cancer via surface GRP78 may antagonize the
cytoprotective UPR.
TM, an ER stress inducer, markedly upregulated GRP78
expression and in 24 h the upregulation peaked. Furthermore,
cultured breast cancer cells did not undergo significant apoptosis
when submitted to extreme degrees of ER stress induced by TM, but
the same treatment induced significant apoptosis in normal
epithelial cells. Obviously, TM aggravated ER stress in cancer
cells and further activated UPR to protect cells against damage
from stress, thus causing ER stress-induced apoptosis. GRP78
expression may serve as a biomarker for activation of the UPR and
play anti-apoptotic properties of the UPR in breast cancer. This
could explain why TM did not induce significant apoptosis in breast
cancer cells, differentiating from normal cells. It is indicated
that the cyto-protective UPR is the base of tumor resistance and
GRP78 plays a central role in the process.
The MAPK pathway is known to be activated in a broad
spectrum of human tumors, including human colorectal (25) and gastric cancer (26) and melanoma (27). We observed that activation of the
MEK/ERK pathway was relatively weak in breast cancer cells, yet TM
induced constitutive activation of the pathway as evidenced by
increased phosphorylation (activation) form of ERK1/2 (P-ERK1/2).
The MEK inhibitor was found to downregulate GRP78 expression and
block activation of the UPR induced by TM. This indicates that
activation of the MEK/ERK pathway plays an important role in
upregulation of GRP78 by ER stress. In light of these results, we
consider that the MEK inhibitor functions upstream of the
activation of UPR and inhibiting the activation of survival pathway
MEK/ERK blocks the cyto-protective role of the UPR. It was
confirmed by ER stress-induced apoptosis, which is readily
triggered when the MEK/ERK pathway is inhibited by the MEK
inhibitor U0126. We also found that the MEK inhibitor alone did not
cause appreciable apoptotic cell death in breast cancer cells.
Therefore the MEK/ERK pathway inhibition would be responsible for
the increasing sensitivity of ER stress-induced apoptosis.
Furthermore, the ER chaperon GRP78 seems to be a target of the
MEK/ERK pathway responsible for the inhibition of ER stress-induced
apoptosis. We consider that inhibiting GRP78 expression may enhance
ER stress-induced apoptosis.
GRP78 expression is primarily regulated at the
transcriptional level, mediated by multiple copies of the ER stress
response element within the GRP78 promoter region (28). We showed by RT-qPCR that GRP78 mRNA
levels were decreased by inhibition of MEK/ERK in the presence or
absence of TM, indicating that the MEK/ERK pathway may participate
in regulation of GRP78 transcription. GRP78 is induced in a wide
variety of cancer cells and cancer biopsy tissue. The
identification of the transcription factors interacting with the ER
stress response element leads to the discovery of multiple pathways
whereby mammalian cells can sense ER stress and trigger the
transcription of GRP78 (29). We
utilized GRP78 knockdown by siRNA resulting in significant
increases in sensitivity of breast cancer cells to TM-induced
apoptosis, but GRP78 siRNA alone, without TM induction, resulted in
only slight increase in apoptosis. This suggests that GRP78 seems
to be a target of ER stress-induced apoptosis which it appears
anti-apoptotic only when ER stress is induced.
We also found that sensitization of breast cancer
cells to TM-induced apoptosis by inhibition of MEK/ERK was
caspase-4 dependent, in that when TM induced ER stress, caspase-4
activity was lower, yet MEK/ERK inhibition and GRP78 knockdown
increased levels of caspase-4 activity. Caspase-4 is an apical
caspase in induction of apoptosis by TM in the presence of U0126,
activation of caspase-4 necessarily switched on caspase cascade
responses to apoptosis. Moreover, inhibition of GRP78 by siRNA
enhanced activation of caspase-4 induced by TM. These results
indicate that GRP78 may participate in controlling the activation
of caspase-4 in breast cancer cells.
In summary, GRP78 knockdown resulted in significant
increases in sensitivity of cells to apoptosis by TM. Inhibition of
MEK/ERK may sensitize breast cancer cells to TM-induced apoptosis.
The results indicate that it may be an effective strategy against
breast cancer to combine agents that induce ER stress with those
inhibiting the MEK/ERK pathway. However, normal cells WI-38 and
MCF-10A seem to be relatively sensitive to TM-induced apoptosis,
indicating that careful evaluation of clinically relevant ER
stress-inducing agents in combination with inhibitors targeting the
MEK/ERK pathway is required before in vivo investigations
being carried out. Of interest, GRP78 is found to be expressed at
relatively high levels in cultured breast cancer cells and breast
cancer tissues, but is hardly detectable in WI-38, MCF-10A and
breast fibroadenoma. This suggests that targeting GRP78 may have
tumor-specific selectivity and thus being useful in treatment of
breast cancer, this role is due, at least partly, to activation of
caspase-4. GRP78 may be a potential target against breast cancer,
and small molecular inhibitor targeting GRP78 in combination with
those inducing ER stress, is a promising chemotherapeutical plan
against breast cancer.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (31401094).
References
|
1
|
Pluquet O, Pourtier A and Abbadie C: The
unfolded protein response and cellular senescence. A review in the
theme: Cellular mechanisms of endoplasmic reticulum stress
signaling in health and disease. Am J Physiol Cell Physiol.
308:C415–C425. 2015. View Article : Google Scholar
|
|
2
|
Manié SN, Lebeau J and Chevet E: Cellular
mechanisms of endoplasmic reticulum stress signaling in health and
disease. 3 Orchestrating the unfolded protein response in
oncogenesis: An update. Am J Physiol Cell Physiol. 307:C901–C907.
2014. View Article : Google Scholar
|
|
3
|
Bian Q, Fernandes AF, Taylor A, Wu M,
Pereira P and Shang F: Expression of K6W-ubiquitin in lens
epithelial cells leads to upregulation of a broad spectrum of
molecular chaperones. Mol Vis. 14:403–412. 2008.PubMed/NCBI
|
|
4
|
Shoulders MD, Ryno LM, Genereux JC,
Moresco JJ, Tu PG, Wu C, Yates JR III, Su AI, Kelly JW and Wiseman
RL: Stress-independent activation of XBP1s and/or ATF6 reveals
three functionally diverse ER proteostasis environments. Cell Rep.
3:1279–1292. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ni M, Lee AS and Ni MandLee AS: ER
chaperones in mammalian development and human diseases. FEBS Lett.
581:3641–3651. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jaronen M, Goldsteins G and Koistinaho J:
ER stress and unfolded protein response in amyotrophic lateral
sclerosis-a controversial role of protein disulphide isomerase.
Front Cell Neurosci. 8:4022014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rainbolt TK, Saunders JM and Wiseman RL:
Stress-responsive regulation of mitochondria through the ER
unfolded protein response. Trends Endocrinol Metab. 25:528–537.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang M, Wey S, Zhang Y, Ye R and Lee AS:
Role of the unfolded protein response regulator GRP78/BiP in
development, cancer, and neurological disorders. Antioxid Redox
Signal. 11:2307–2316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsuo K, Gray MJ, Yang DY, Srivastava SA,
Tripathi PB, Sonoda LA, Yoo EJ, Dubeau L, Lee AS and Lin YG: The
endoplasmic reticulum stress marker, glucose-regulated protein-78
(GRP78) in visceral adipocytes predicts endometrial cancer
progression and patient survival. Gynecol Oncol. 128:552–559. 2013.
View Article : Google Scholar
|
|
10
|
Schröder M and Kaufman RJ: ER stress and
the unfolded protein response. Mutat Res. 569:29–63. 2005.
View Article : Google Scholar
|
|
11
|
Wang C, Jiang K, Gao D, Kang X, Sun C,
Zhang Q, Li Y, Sun L, Zhang S, Guo K, et al: Clusterin protects
hepatocellular carcinoma cells from endoplasmic reticulum stress
induced apoptosis through GRP78. PLoS One. 8:e559812013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pootrakul L, Datar RH, Shi SR, Cai J,
Hawes D, Groshen SG, Lee AS and Cote RJ: Expression of stress
response protein Grp78 is associated with the development of
castration-resistant prostate cancer. Clin Cancer Res.
12:5987–5993. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mhaidat NM, Wang Y, Kiejda KA, Zhang XD
and Hersey P: Docetaxel-induced apoptosis in melanoma cells is
dependent on activation of caspase-2. Mol Cancer Ther. 6:752–761.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choudhury S, Bhootada Y, Gorbatyuk O and
Gorbatyuk M: Caspase-7 ablation modulates UPR, reprograms TRAF2-JNK
apoptosis and protects T17M rhodopsin mice from severe retinal
degeneration. Cell Death Dis. 4:e5282013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Estornes Y, Aguileta MA, Dubuisson C, De
Keyser J, Goossens V, Kersse K, Samali A, Vandenabeele P and
Bertrand MJ: RIPK1 promotes death receptor-independent
caspase-8-mediated apoptosis under unresolved ER stress conditions.
Cell Death Dis. 6:e17982015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Banerjee C, Singh A, Das TK, Raman R,
Shrivastava A and Mazumder S: Ameliorating ER-stress attenuates
Aeromonas hydrophila-induced mitochondrial dysfunctioning and
caspase mediated HKM apoptosis in Clarias batrachus. Sci Rep.
4:58202014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sandow JJ, Dorstyn L, O'Reilly LA, Tailler
M, Kumar S, Strasser A and Ekert PG: ER stress does not cause
upregulation and activation of caspase-2 to initiate apoptosis.
Cell Death Differ. 21:475–480. 2014. View Article : Google Scholar :
|
|
18
|
Binet F, Chiasson S and Girard D: Evidence
that endoplasmic reticulum (ER) stress and caspase-4 activation
occur in human neutrophils. Biochem Biophys Res Commun. 391:18–23.
2010. View Article : Google Scholar
|
|
19
|
Matsuzaki S, Hiratsuka T, Kuwahara R,
Katayama T and Tohyama M: Caspase-4 is partially cleaved by calpain
via the impairment of Ca2+ homeostasis under the ER
stress. Neurochem Int. 56:352–356. 2010. View Article : Google Scholar
|
|
20
|
Adachi T, Teramachi M, Yasuda H, Kamiya T
and Hara H: Contribution of p38 MAPK, NF-κB and glucocorticoid
signaling pathways to ER stress-induced increase in retinal
endothelial permeability. Arch Biochem Biophys. 520:30–35. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Feng R, Zhai WL, Yang HY, Jin H and Zhang
QX: Induction of ER stress protects gastric cancer cells against
apoptosis induced by cisplatin and doxorubicin through activation
of p38 MAPK. Biochem Biophys Res Commun. 406:299–304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin ML, Chen SS, Huang RY, Lu YC, Liao YR,
Reddy MV, Lee CC and Wu TS: Suppression of PI3K/Akt signaling by
synthetic bichalcone analog TSWU-CD4 induces ER stress-and
Bax/Bak-mediated apoptosis of cancer cells. Apoptosis.
19:1637–1653. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Z, Zhang H, Xu X, Shi H, Yu X, Wang
X, Yan Y, Fu X, Hu H, Li X, et al: bFGF inhibits ER stress induced
by ischemic oxidative injury via activation of the PI3K/Akt and
ERK1/2 pathways. Toxicol Lett. 212:137–146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee AS: The ER chaperone and signaling
regulator GRP78/BiP as a monitor of endoplasmic reticulum stress.
Methods. 35:373–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mhaidat NM, Alali FQ, Matalqah SM, Matalka
II, Jaradat SA, Al-Sawalha NA and Thorne RF: Inhibition of MEK
sensitizes paclitaxel-induced apoptosis of human colorectal cancer
cells by downregulation of GRP78. Anticancer Drugs. 20:601–606.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kang W, Tong JH, Chan AW, Lee TL, Lung RW,
Leung PP, So KK, Wu K, Fan D, Yu J, et al: Yes-associated protein 1
exhibits oncogenic property in gastric cancer and its nuclear
accumulation associates with poor prognosis. Clin Cancer Res.
17:2130–2139. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oh YT, Deng J, Yue P, Owonikoko TK, Khuri
FR and Sun SY, Khuri FR and Sun SY: Inhibition of B-Raf/MEK/ERK
signaling suppresses DR5 expression and impairs response of cancer
cells to DR5-mediated apoptosis and T cell-induced killing.
Oncogene. Apr 13–2015.Epub ahead of print. View Article : Google Scholar
|
|
28
|
Tang J, Guo YS, Zhang Y, Yu XL, Li L,
Huang W, Li Y, Chen B, Jiang JL and Chen ZN: CD147 induces UPR to
inhibit apoptosis and chemosensitivity by increasing the
transcription of Bip in hepatocellular carcinoma. Cell Death
Differ. 19:1779–1790. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J and Lee AS: Stress induction of
GRP78/BiP and its role in cancer. Curr Mol Med. 6:45–54. 2006.
View Article : Google Scholar : PubMed/NCBI
|