Introduction
Glioma is the most common primary intracranial
tumor, accounting for 35.26–60% of human intracranial tumors and
including up to 60% of malignant neuroastrocytomas, leading to poor
prognosis and high mortality (1).
One characteristic of glioma is that glioma cells often invade
important functional areas of the normal brain tissue; these
'satellite lesions' surrounding the primary lesion are formed
around the primary lesion (2–6).
Therefore, simple excision always presents obvious limitations
between ensuring cerebral function and removing the tumor fractions
(7,8). Since glioma pathogenesis has not been
fully defined, the discussion regarding its etiology, pathogenesis,
biological characteristics and new effective treatments has become
a hot topic in neurosurgery.
SEPT7 is a member of the septins family with GTPase
activity, and is also a kind of cytoskeletal protein that is highly
expressed in the central nervous system (9,10).
Since the physiological functions and biochemical characteristics
of SEPT7 protein were discovered, the results have shown that SEPT7
is involved not only in the cytoskeleton and participates in the
regulation of cytokinesis, but is also involved in various
diseases. As a consequence, the relationship between SEPT7
expression and human glioma has gradually become a concern. The
study by Jia et al (9)
showed that the overexpression of SEPT7 can inhibit cell
proliferation and cell cycle arrest in the G0/G1 phase, further
suppressing glioma cell growth both in vitro and in
vivo. Suppressing the tumor-suppressor gene SEPT7 can promote
glioblastoma cell migration and invasion, and it was verified that
SEPT7 affected remodeling of the actin cytoskeleton in glioblastoma
cells (11). Upregulation of SEPT7
gene can inhibit cell invasion, and downregulate the expression of
MMP2/9, MT1-MMP, and integrin α(v)β(3) (12).
Previous studies have demonstrated that cofilin is
involved in the homeostasis of actin polymerization and severing
(13,14). Cofilin is inactivated by
phosphorylation at Ser-3 by LIMK1/2 and TESK1/2 and reactivated by
dephosphorylation by SSHs, CIN and the other protein phosphatases,
such as PP1 and PP2A (14). Cofilin
activation can prompt actin polymerization, while cofilin
inactivation, on the contrary, can lead to actin severing.
Similarly, SEPT7 has also been shown to be involved in the
migration of actin cytoskeleton in vitro and it is
conceivable that there is a link between SEPT7 and cofilin in
stabilizing actin filaments (15).
Until now, the molecular mechanisms by which SEPT7
impact glioma cell invasion are still unclear. In the present
study, we examined the role of SEPT7 as a cytoskeletal proteins
with GTPase activity, in the rearrangement of the actin
cytoskeleton and tumor cell migration and invasion, to further
examine interactions between SEPT7 and cofilin phosphoregulation in
LN18 glioma cells.
Materials and methods
Cell culture
The human brain glioma cell lines LN229 and LN18
were obtained from the American Type Culture Collection (ATCC,
Manassas, VA, USA), U87 and U251 were obtained from Sigma-Aldrich
(St. Louis, MO, USA) and the normal human brain gliocyte cell line
(HEB) was obtained from Shanghai, China. Cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS), 100 U/ml penicillin, and 100 mg/ml
streptomycin (all from Invitrogen, Carlsbad, CA, USA) and
maintained at 37°C in a humidified incubator of 5%
CO2.
Western blot analysis
Total proteins were extracted using the Tissue or
Cell Total Protein Extraction kit (Amresco, Solon, OH, USA) from
LN229, LN18, U87 and U251 cell lines. All of the primary antibodies
were purchased from Abcam (Cambridge, UK). The proteins were
separated by SDS-PAGE followed by electrotransfer to an NC
membrane; the membranes were probed using antibodies against SEPT7
(1:2,000), MMP-2 (1:1,000), MMP-9 (1:1,000), cofilin (1:1,000),
phospho(ser3)-cofilin (1:1,000), and actin (1:5,000), followed by a
horseradish peroxidase (HRP)-conjugated second antibody (GAPDH,
1:10,000) (Abcam). Bands were revealed with ECL reagent (Millipore,
billerica, MA, USA) and recorded on X-ray film (Kodak, Xiamen,
China). The densitometry of each band was quantified by a gel
imaging system and Quantity One 4.62 software (Bio-Rad, Hercules,
CA, USA).
RT-PCR
Total RNA was extracted using TRIzol reagents
(Invitrogen) from LN229, LN18, U87 and U251 cell lines. Isolated
RNA was electrophoresed on 1% agarose gel to detect the purity of
total RNA. The first-strand cDNA was synthesized using 1 µg
total RNA and SuperScript® III Reverse Transcriptase
(Invitrogen). PCR amplification was performed using the PCR
amplification kit (Takara Bio, Inc., Otsu, Japan). The specific
primers were designed using Primer Premier 6.0 software and
synthesized by Sangon biotech (Shanghai, China). The primers for
SEPT7 were 5′-CTCTTGCTGTGGTAGGTAG-3′ (forward) and
5′-GCTTCTGTAGTTCTCATAGTG-3′ (reverse). The primers for GAPDH as an
internal control were 5′-GAGTGAGTGGAAGACAGAAT-3′ (forward) and
5′-GCAGAGAAGCAGACAGTTA-3′ (reverse).
The SEPT7 overexpression vector and siRNA
transfection
The pcDNA3.1(+)/SEPT7 expression vector was
constructed by cloning SEPT7 fragment from normal human cDNA into
pcDNA3.1(+) (Invitrogen) between BamHI and EcoRI
sites to express SEPT7 in abundance in E. coli DH5α cells.
The primers for SEPT7 were as follows: forward primer,
5′-GGAGGATCCCTCTTGCTGTGGTAGGTAG-3′ (including BamHI site
GGATCC and three protection bases) and reverse primer,
5′-CTCGAATTCGCTTCTGTAGTTCTCATAGTG-3′ (including EcoRI site
GGATCC and three protection bases). The recombinant plasmid was
identified by endonuclease digestion and DNA sequencing. The
pcDNA3.1(+)/SEPT7 and pcDNA3.1(+) plasmids were separately
transfected into the glioma cells mediated by Lipofectamine 2000
(Invitrogen) as per the manual. The stably transfected clones were
screened by G418, and identified by western blot analysis and
RT-PCR. Finally, the siRNA or control siRNA was also transfected
into the glioma cells mediated by Lipofectamine 2000 (Invitrogen)
as manual. Within 24 h after transfection, the protein level was
detected by western blot analysis and RT-PCR. The siRNA primers
sequences were designed by Invitrogen Block-iT RNAi designer.
Transwell migration and cell chemotaxis
assay
The cells were added into Transwell chambers
(Millipore) and FBS was added into lower chamber. Cells were
cultured at 37°C for 24 h in a humidified incubator of 5%
CO2, and then the hematoxylin and eosin (H&E)
stained cells in the lower chamber were counted under the
microscope. The cells in 10 high power fields were randomly chosen
and the average transmembrane cell number per high-power field was
shown as cell migration ability. For the cell chemotaxis assay, the
different concentrations of IGF-1 (0, 1, 10 and 100 ng/ml) were
added into the lower chamber of chemotaxis chambers (Millpore) with
30 µl per well. There were membranes with 8 µm gaps
between the upper and lower chambers. Then, 50 µl of cell
suspension with was added into the upper chamber of each well.
After cells were cultured at 37°C for 24 h, the H&E stained
cells in the lower chamber were counted under the microscope. The
chemotactic index was presented as cell migration number under
IGF-1 and without IGF-1.
F-actin/G-actin ratio determination
The cells were broken, homogenized in cold lysis
buffer (10 mM K2HPO4, 100 mM NaF, 50 mM KCl,
2 mM MgCl2, 1 mM EgTA, 0.2 mM DTT, 0.5% Triton X-100, 1
mM sucrose, pH 7.0) and centrifuged at 15,000 × g for 30 min.
Soluble actin (G-actin) was measured in the supernatant. The
insoluble F-actin in the pellet was resuspended in lysis buffer
plus an equal volume of buffer 2 (1.5 mM guanidine hydrochloride, 1
mM sodium acetate, 1 mM CaCl2, 1 mM ATP and 20 mM
Tris-HCl, pH 7.5) and incubated on ice for 1 h to convert F-actin
into soluble G-actin, with gentle mixing every 15 min. The samples
were centrifuged at 15,000 × g for 30 min, and F-actin was measured
in this supernatant. Samples from the supernatant (G-actin) and
pellet (F-actin) fractions were proportionally loaded and analyzed
by western blot analysis using a specific actin antibody
(Millipore, Temecula, CA, USA).
Statistical analysis
Data are presented as means ± SD. Statistical
analysis was performed with SPSS13.0 software (IBM, Armonk, NY,
USA). Statistical evaluation of the data was performed using
one-way ANOVA and LSD for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
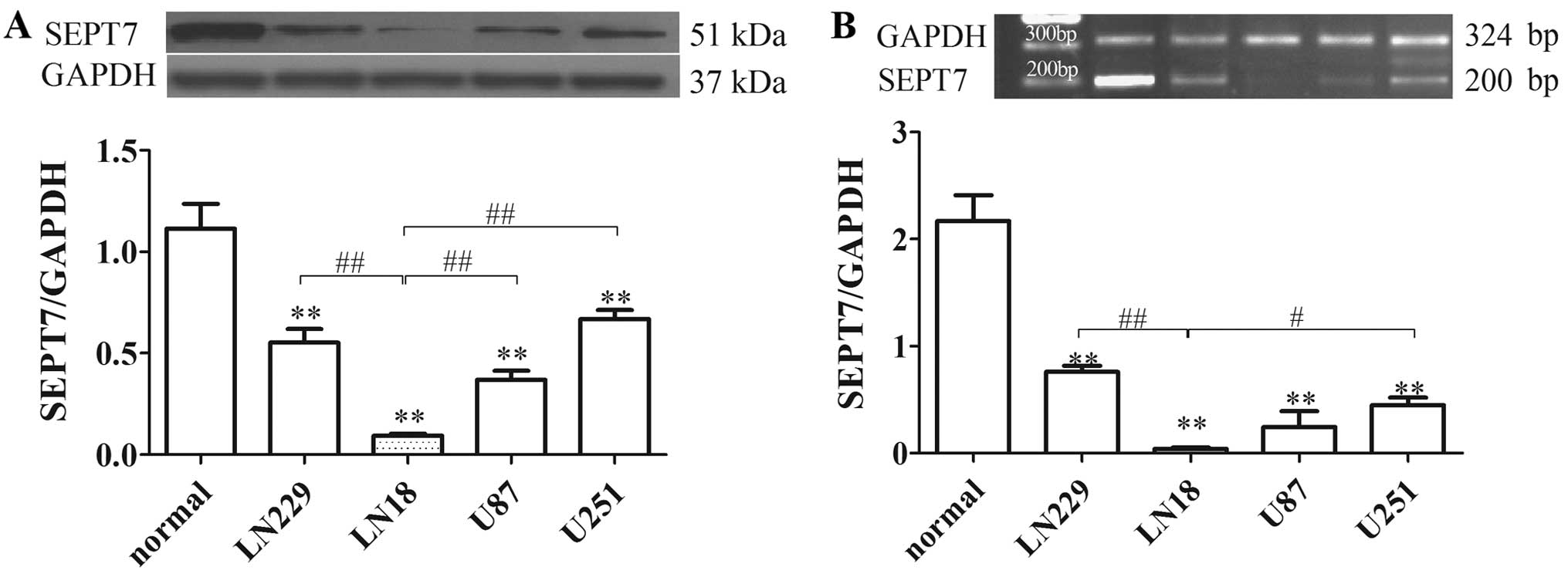
SEPT7 is downregulated in human brain
glioma cells
SEPT7 expression was identified by western blot
analysis and RT-PCR in various glioma cells. The results showed
that SEPT7 levels in glioma cells were reduced by 50% or less
compared to normal human brain cells. SEPT7 levels in LN18 cell
lines were lower than in other glioma cells (Fig. 1). LN18 was derived from a patient
with a grade IV glioma in the right temporal lobe (ATCC). The
subsequent tests therefore used the LN18 cell line as the research
object.
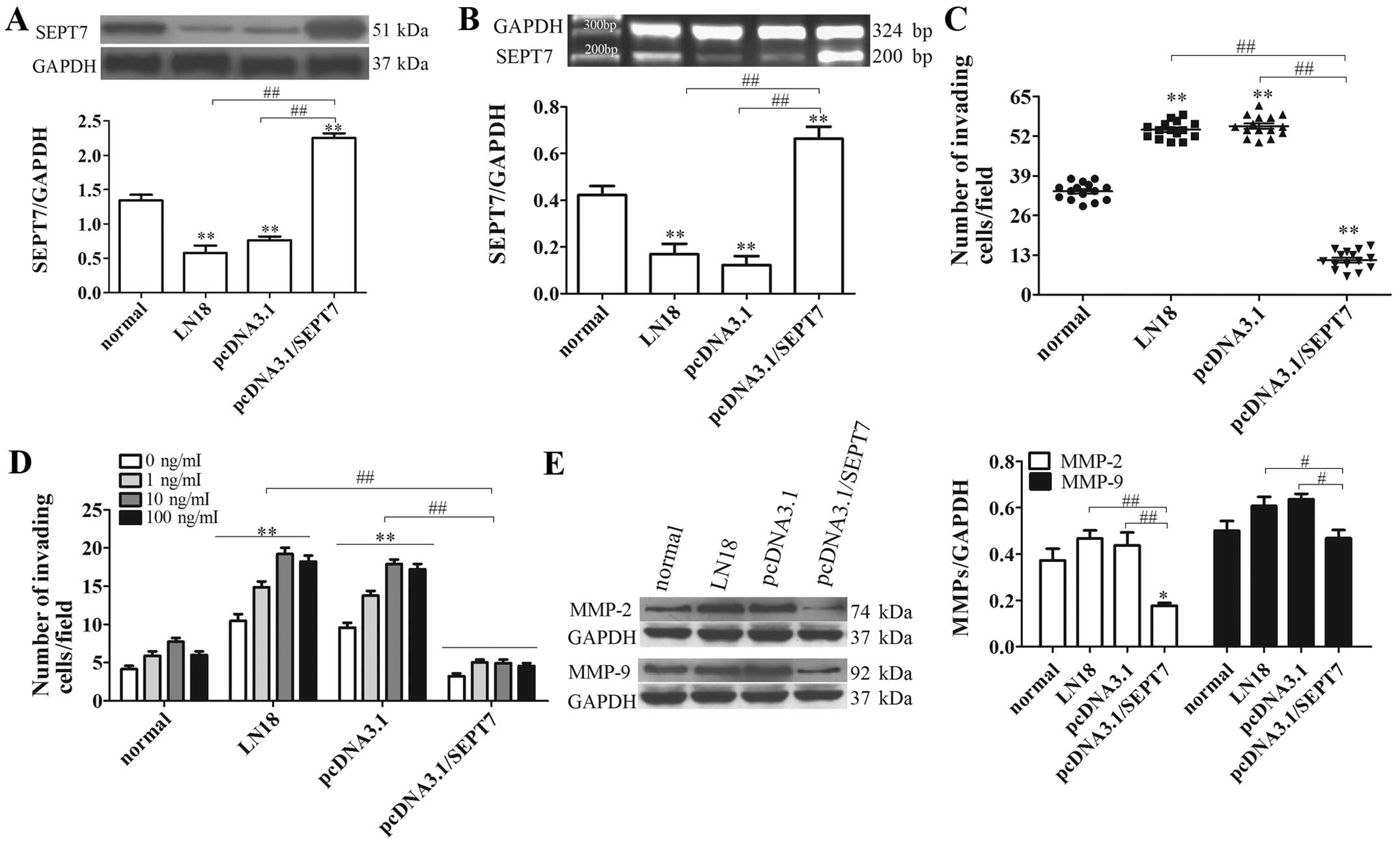
SEPT7 overexpression suppresses human
brain glioma cell migration
To verify whether SEPT7 influences glioma invasion,
a SEPT7 overexpression vector was constructed and transfected into
the LN18 cell line (Fig. 2A and B).
Analysis of SEPT7 expression in LN18 cells after transfection with
SEPT7 overexpression plasmid indicated that the level of SEPT7 in
pcDNA3.1/SEPT7 had a marked elevation compared to LN18 (P<0.01)
(Fig. 2A and B). Malignant tumor
invasion and metastasis mainly depend on cell migration, as well as
extracellular matrix degradation and the induction of chemokines.
Given the increased SEPT7 levels in pcDNA3.1/SEPT7, we examined
whether a redundancy in SEPT7 alone accounts for the failure of
glioma cells to invade and transfer. The results showed that the
number of invading cells in the pcDNA3.1/SEPT7 group were
significantly reduced in contrast to the LN18 group (P<0.01)
(Fig. 2C), indicating that SEPT7
made no contribution to tumor cell migration. The effect of SEPT7
on tumor cell invasion and metastasis was further studied using an
IGF-1-induced cell chemotaxis assay. Under the treatment of IGF-1
at different concentrations, glioma cell chemotaxis had a
significant difference; overall, 10 ng/ml IGF-1 was the most
powerful for glioma cell chemotaxis, either in LN18 or
pcDNA3.1/SEPT7 (Fig. 2D).
Consistent with cell migration results, the chemotaxis assay
revealed that SEPT7 overexpression markedly diminished IGF-1
induced tumor cell chemotaxis compared to LN18 (P<0.01)
(Fig. 2D). These results indicated
that increased SEPT7 inhibits glioma cell migration and
chemotaxis.
Activation of MMP-2 and MMP-9 regulates both matrix
degradation and motility, thereby facilitating cellular invasion
(16,17). The levels of MMP-2 and MMP-9 were
probed with western blot analysis, indicating that the levels of
MMP-2 and MMP-9 in pcDNA3.1/SEPT7 were markedly depressed,
respectively, compared to LN18 (P<0.01 and P<0.05) (Fig. 2E). These results suggest that
increased SEPT7 suppresses extracellular matrix degradation,
consequently opposing cellular invasion.
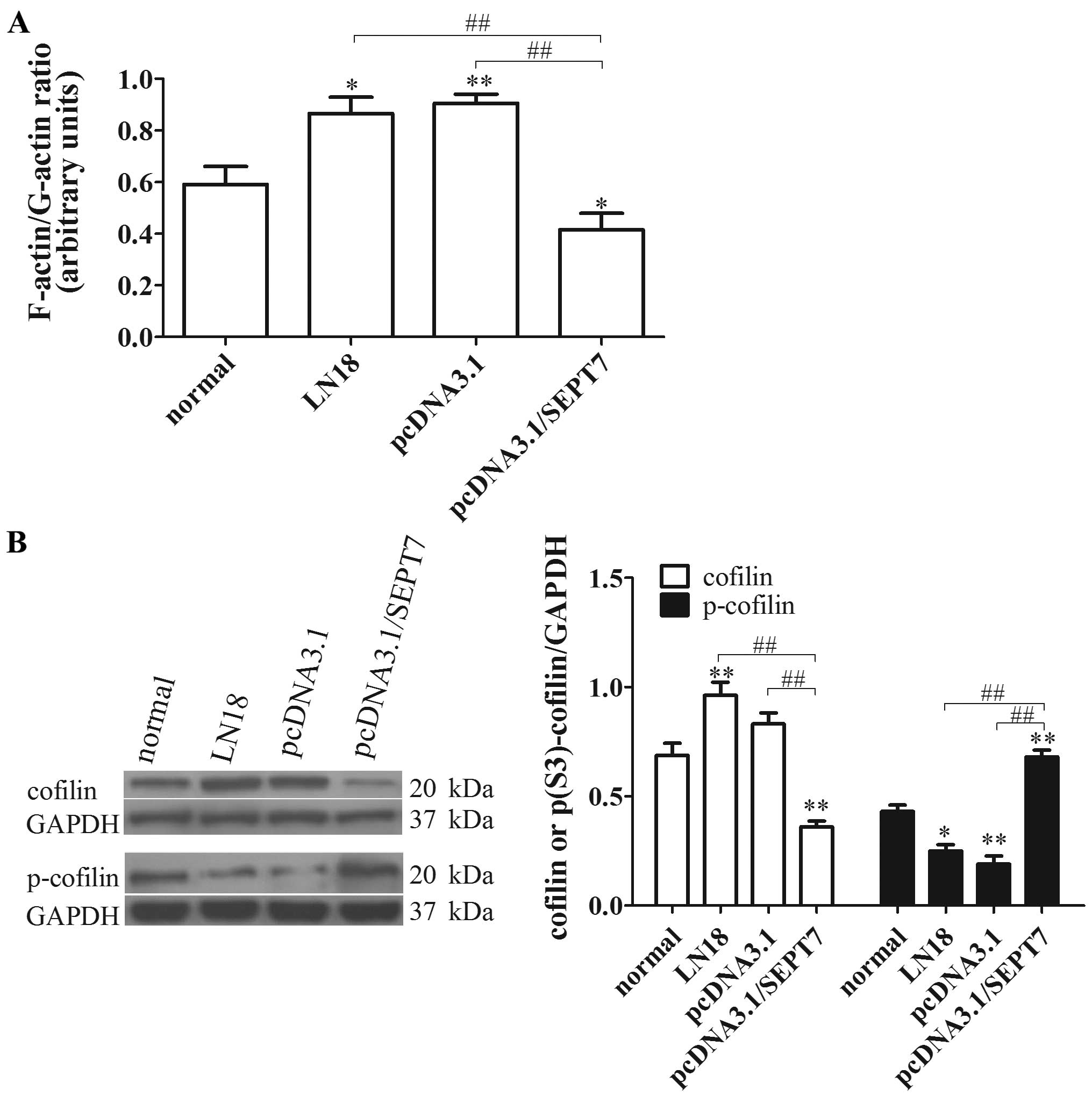
SEPT7 overexpression inhibits
cytoskeleton locomotion in human glioma cells
SEPT7 is a cytoskeletal protein and is involved in
the entry of axonal microtubules into nascent filopodia, enabling
the formation of collateral branches (18). Increased SEPT7 binds to the actin
filaments and promotes F-actin ring formation, against cell
migration (19). Consistent with
this conclusion, our study indicated that the F-actin/G-actin ratio
in pcDNA3.1/SEPT7 was significantly reduced compared with the LN18
group (P<0.01) (Fig. 3A),
revealing that SEPT7 upregulation can promote actin
depolymerization.
Cofilin also acts as an actin-binding protein that
plays an essential role in regulating actin filament dynamics and
reorganization by stimulating the severance and depolymerization of
actin filaments (14). Therefore,
we speculated that a possible relationship exists between SEPT7 and
cofilin phospho-regulation to modulate actin filament dynamics. To
verify this conjecture, the levels of cofilin and p-cofilin were
tested; the results showed that cofilin level in the pcDNA3.1/SEPT7
group decreased visibly compared with the LN18 group (P<0.01),
while the p-cofilin level showed the opposite results (P<0.01)
(Fig. 3B). These findings
preliminary clarified that increased SEPT7 promotes the
depolymerization of actin filaments, and is concerned with cofilin
phospho-regulation.
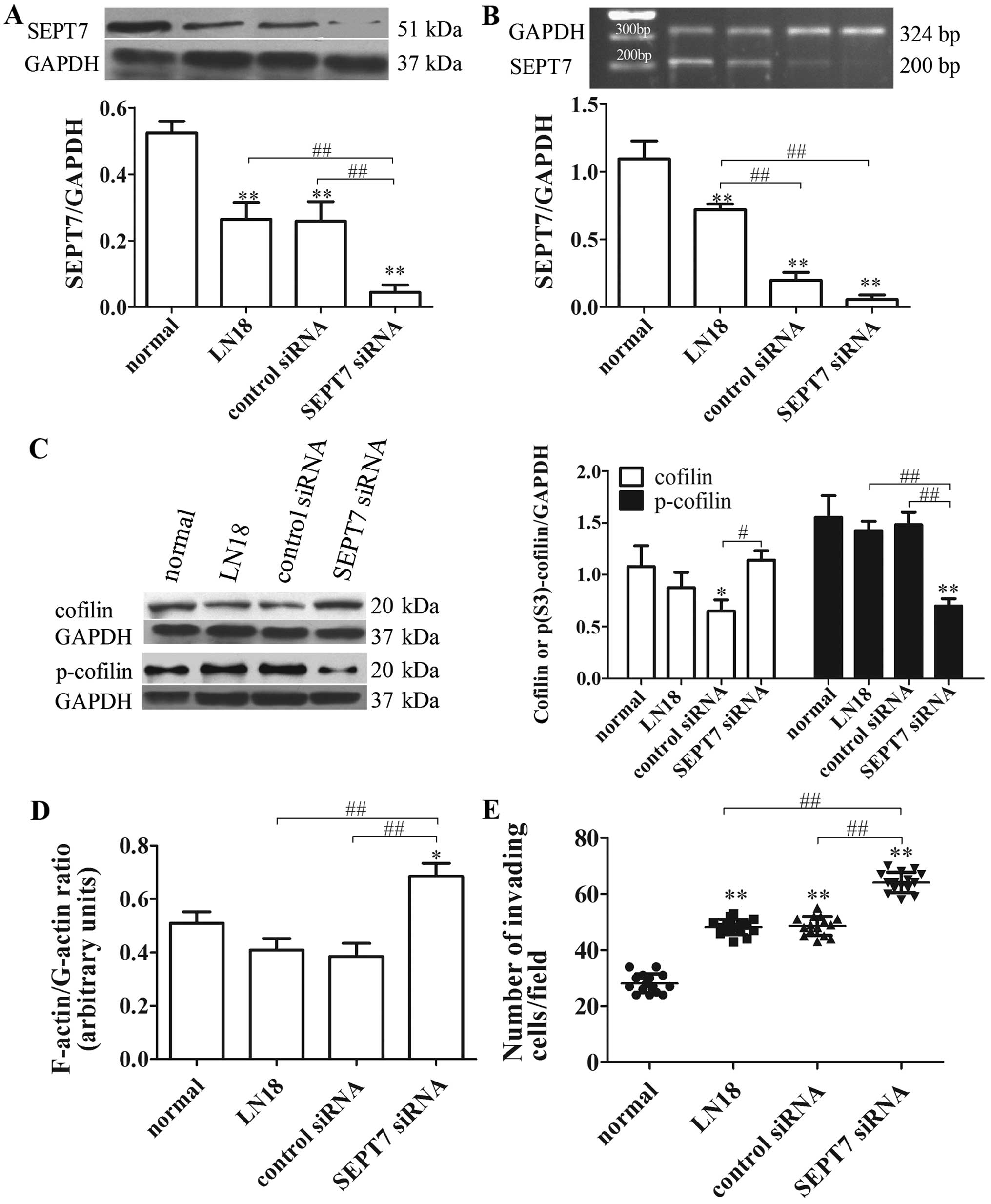
SEPT7 siRNA increases cell migration and
cytoskeleton locomotion in human glioma cells
To further explore the roles of cytoskeleton
signaling in reduced SEPT7-mediated glioma cell migration, the
control siRNA and SEPT7 siRNA were transfected into the LN18 cells.
The degrees of transfection were tested at the protein and nucleic
acid levels, revealing that the amount of SEPT7 was markedly
reduced compared with the LN18 group (P<0.01 and P<0.01)
(Fig. 4A and B). Cofilin,
F-actin/G-actin ratio and cell migration in the SEPT7 siRNA group
were significantly improved compared with the LN18 group
(P<0.05, P<0.01 and P<0.01) (Fig. 4C–E), while p-cofilin expression
reduced (P<0.01) (Fig. 4C).
Thus, these experiments show that the knockdown of SEPT7 enhances
the motility of glioma cells through cofilin phospho-regulation,
which accelerates actin polymerization.
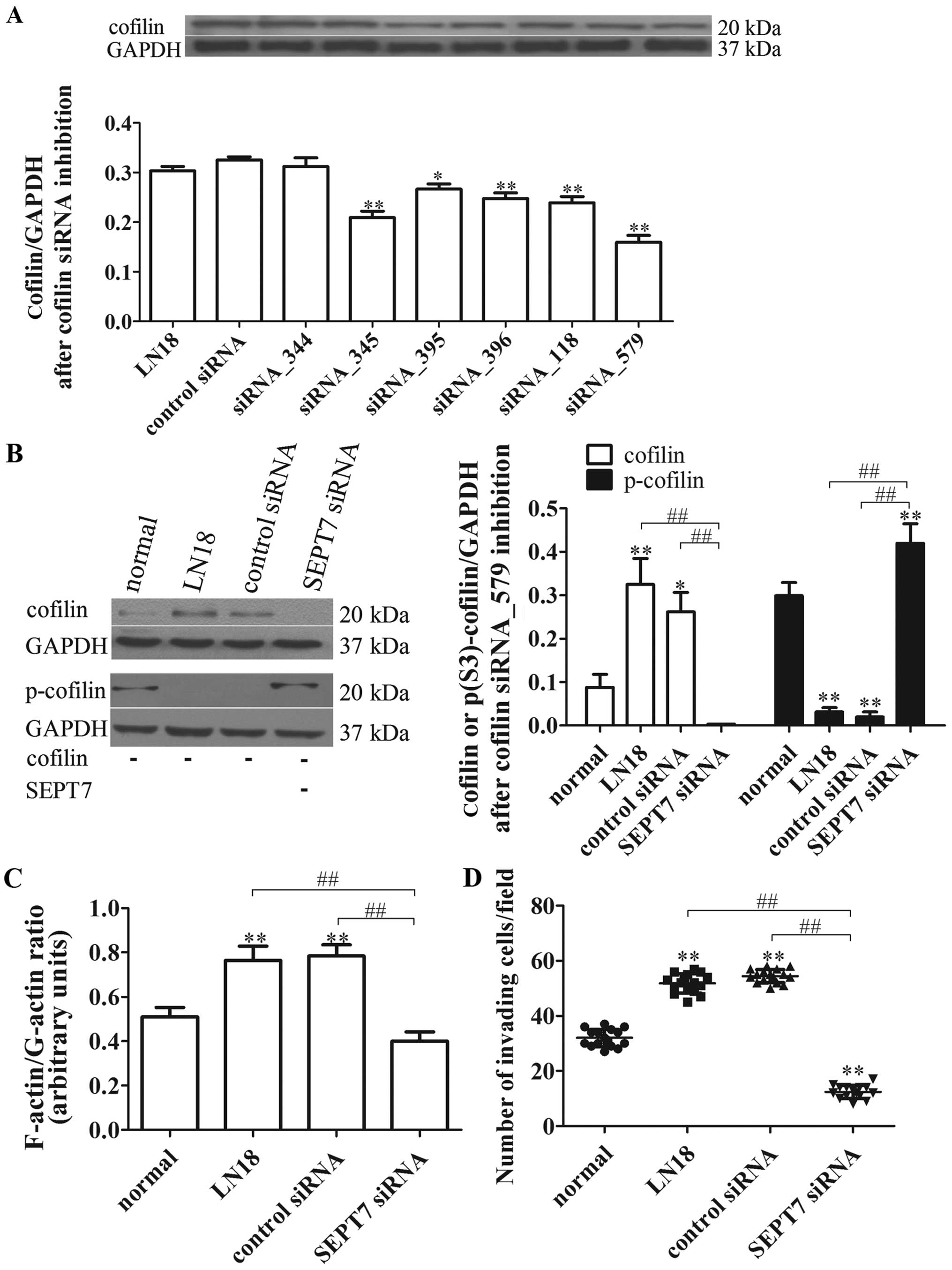
Cofilin inhibition suppresses SEPT7
siRNA-induced cell migration
In view of the critical roles of SEPT7 and cofilin
in linking signaling pathways to actin cytoskeleton remodeling, we
further tested whether cofilin is involved in the process of SEPT7
siRNA-induced actin aggregation. The different cofilin siRNA were
designed and transfected, respectively, into the LN18 cell lines,
and the degrees of transfection were tested, showing that cofilin
siRNA_579 was the best for inhibiting cofilin protein expression
(P<0.01) (Fig. 5A). Afterwards,
the cofilin siRNA_579 was transfected, respectively, into the
normal, LN18, control siRNA and SEPT7 siRNA group cells. The cell
migration and cytoskeleton locomotion were measured; as shown in
Fig. 5B, the levels of cofilin and
p-cofilin after cofilin inhibition were reduced in general in all
groups compared with unrestrained cofilin (Figs. 4C and 5B). However, the level of cofilin in the
SEPT7 siRNA group was further reduced compared with the LN18 group
(P<0.01), while the p-cofilin level was just the opposite
(P<0.01) (Fig. 5B).
Importantly, the F-actin/G-actin ratio and cell
migration under cofilin inhibition in SEPT7 siRNA group was
remarkably declined compared with the LN18 group (P<0.01 and
P<0.01) (Fig. 5C and D). These
findings suggest that actin polymerization and cell migration of
SEPT7 siRNA-induced are reversed by cofilin inhibition and are
cofilin-dependent.
Discussion
In the past two decades, although there has been a
series of active comprehensive treatment of glioma developed in
clinical practice, the effect is still not ideal as tumor cells
find it easy to migrate and are difficult to control. Therefore,
human gliomas are still considered a refractory disease in the
neurosurgery field. As a result, it is necessary to determine the
molecular mechanism of glioma cell locomotion and invasion.
The interstitial movement is encephalic motion in
glioma cells; tumor cells in the subependymal zone and soft
membrane area can migrate via an amoeboid movement (20,21).
During cell migration, the formation of front-end protuberance
lamellipodia and filopodia plays an important role, and
lamellipodia are considered to be the main driver of cell motility
(22,23). However, lamellipodia are formed
during actin polymerization and assemble in the front-end
protuberance of lamellipodia (24).
Consistently, our analyses suggested that actin polymerization
accelerates cell migration through cytoskeleton signaling molecules
and actin-binding proteins.
SEPT7 has been identified in all eukaryocytes. The
study by Jia et al (9)
observed that SEPT7 expression was much lower in high-grade than
low-grade gliomas. SEPT7 gene expression was negatively correlated
with the ascending order of glioma grades. It has also been
confirmed that SEPT7 expression in different glioma cells is
significantly different. Currently, most research on SEPT7 is
performed at the cellular level, such as the relationship between
SEPT7 and cell growth and the cell cycle. Our study also found that
SEPT7 overexpression can inhibit cell migration and increase
extracellular matrix degradation. However, our focus was on the
interplay and molecular mechanism responsible for SEPT7 modulating
the cytoskeleton locomotion and cell migration (Fig. 6).
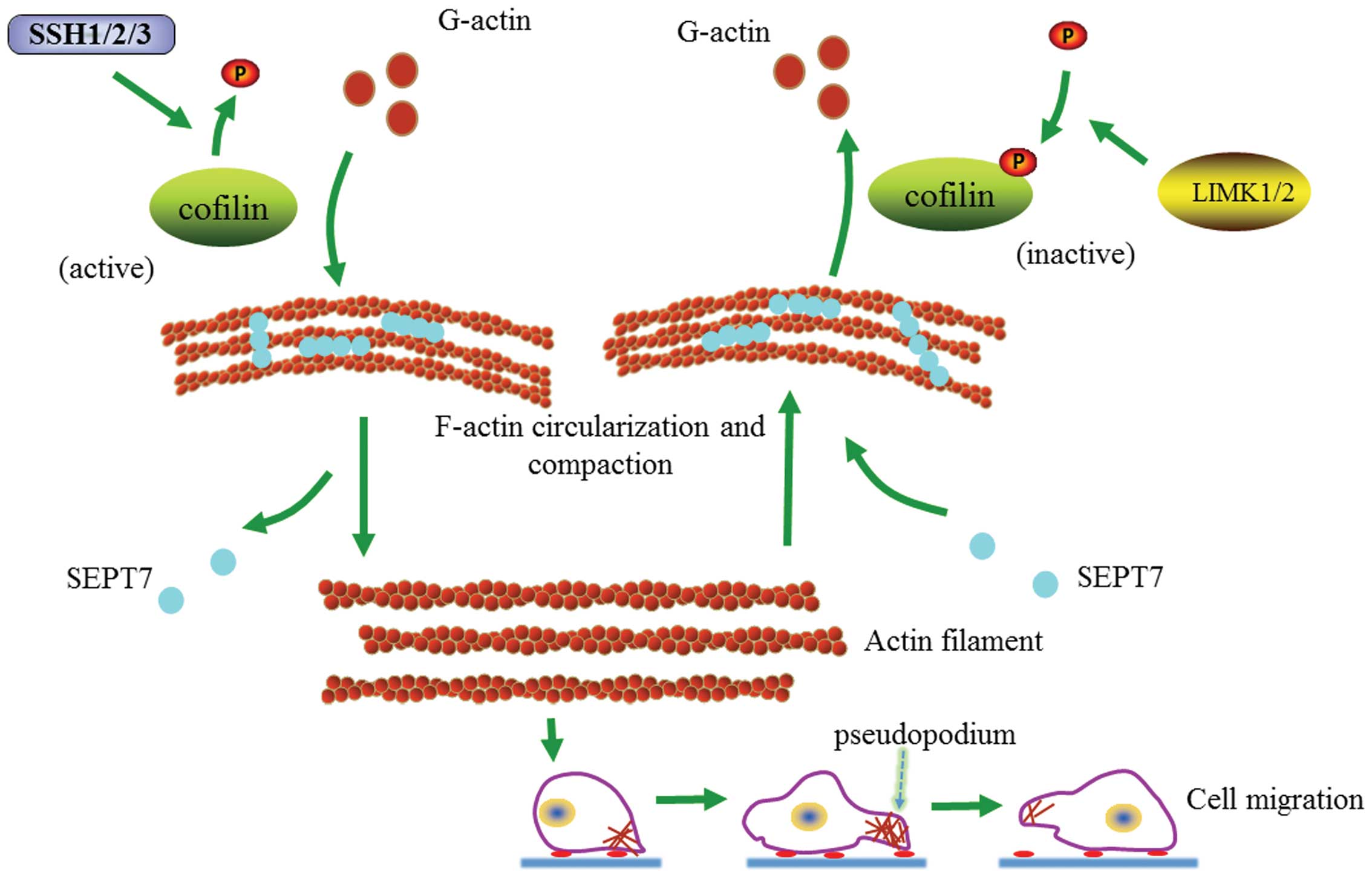 | Figure 6Proposed pathway to expound the
interaction among SEPT7, cofilin phospho-regulation and actin
dynamic equilibrium. The proposed pathway was drawn based on our
research. On the one hand, improved SEPT7 monomers form additional
SEPT7 rods and bind to actin filaments making them bent or
cyclized, before subsequently recruiting phosphorylated cofilin and
binding to actin filaments; cofilin is inactivated by
phosphorylation using LIMK1/2 and urge actin severing. On the other
hand, dissociated SEPT7 from bent or cyclized actin filaments cause
SEPT7 rods to diminish and drop, and SEPT7 monomers that have
detached from the actin filaments can help activated cofilin, which
was dephosphorylated by SSH1/2/3 fast binding to the actin
filaments, causing actin polymerization and accelerating cell
migration. At this time, when cofilin is inhibited, cofilin binding
to actin filaments and SEPT7 reduction decrease, causing slower
cell migration. |
Recent studies have shown that SEPT7 promotes
F-actin ring formation by crosslinking actin filaments into curved
bundles (15). Therefore, SEPT7
could conceivably crosslink with F-actin into loose contractile
networks and SEPT7-mediated actin curving in cells may act in
synergy with myosin-induced actin filament buckling (19,25,26).
Our findings show that increased SEPT7 induces actin
depolymerization and cell migration is consequently hindered. At
the same time, it is accompanied by an increasing cofilin/p-cofilin
ratio. Research has shown that SEPT7 can stabilize actin filaments
bent by cofilin (15). It is
obvious that cofilin and p-cofilin play important roles in the
SEPT7 regulation of actin depolymerization and cell migration. On
the contrary, the downregulation of SEPT7 can increase cell
migration and actin polymerization accompanied by a depressed
cofilin/p-cofilin ratio. Nevertheless, increased actin
polymerization and SEPT7 siRNA-induced cell migration were reversed
when cofilin was inhibited, also accompanied by a change in the
cofilin/p-cofilin ratio. Hence, our study preliminarily suggests
that SEPT7 interacts with cofilin phospho-regulation in modulating
the dynamic equilibrium of actin and cytoskeleton locomotion,
offering a promising candidate for novel therapeutic pathways
against glioma.
References
|
1
|
Ohgaki H and Kleihues P: Epidemiology and
etiology of gliomas. Acta Neuropathol. 109:93–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Claes A, Idema AJ and Wesseling P: Diffuse
Glioma growth: A guerilla war. Acta Neuropathol. 114:443–458. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Giese A and Westphal M: Glioma invasion in
the central nervous system. Neurosurgery. 39:235–250; discussion
250–252. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meyer MA: Malignant gliomas in adults. N
Engl J Med. 359:1850author reply 1850. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mittal S, Szlaczky MC and Barger GR:
Low-grade gliomas in adults. Curr Treat Options Neurol. 10:271–284.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wen PY and Kesari S: Malignant Gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Laws ER, Shaffrey ME, Morris A and
Anderson FA Jr: Surgical management of intracranial gliomas - does
radical resection improve outcome? Acta Neurochir (Suppl).
85:47–53. 2003. View Article : Google Scholar
|
|
8
|
Simon M and Schramm J: Surgical management
of intracranial gliomas. Recent Results Cancer Res. 171:105–124.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jia ZF, Huang Q, Kang CS, Yang WD, Wang
GX, Yu SZ, Jiang H and Pu PY: Overexpression of septin 7 suppresses
glioma cell growth. J Neurooncol. 98:329–340. 2010. View Article : Google Scholar
|
|
10
|
Hall PA, Jung K, Hillan KJ and Russell SE:
Expression profiling the human septin gene family. J Pathol.
206:269–278. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang H, Hua D, Zhang J, Lan Q, Huang Q,
Yoon JG, Han X, Li L, Foltz G, Zheng S, et al: MicroRNA-127-3p
promotes glioblastoma cell migration and invasion by targeting the
tumor-suppressor gene SEPT7. Oncol Rep. 31:2261–2269.
2014.PubMed/NCBI
|
|
12
|
Xu S, Jia ZF, Kang C, Huang Q, Wang G, Liu
X, Zhou X, Xu P and Pu P: Upregulation of SEPT7 gene inhibits
invasion of human glioma cells. Cancer Invest. 28:248–258. 2010.
View Article : Google Scholar
|
|
13
|
Bravo-Cordero JJ, Magalhaes MA, Eddy RJ,
Hodgson L and Condeelis J: Functions of cofilin in cell locomotion
and invasion. Nat Rev Mol Cell Biol. 14:405–415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mizuno K: Signaling mechanisms and
functional roles of cofilin phosphorylation and dephosphorylation.
Cell Signal. 25:457–469. 2013. View Article : Google Scholar
|
|
15
|
Gladfelter AS: Cytoskeleton: Cirque du
septins. Curr Biol. 24:R526–R528. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brooks PC, Strömblad S, Sanders LC, von
Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP and
Cheresh DA: Localization of matrix metalloproteinase MMP-2 to the
surface of invasive cells by interaction with integrin alpha v beta
3. Cell. 85:683–693. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ramos-DeSimone N, Hahn-Dantona E, Sipley
J, Nagase H, French DL and Quigley JP: Activation of matrix
metallopro-teinase-9 (MMP-9) via a converging plasmin/stromelysin-1
cascade enhances tumor cell invasion. J Biol Chem. 274:13066–13076.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu J, Bai X, Bowen JR, Dolat L, Korobova
F, Yu W, Baas PW, Svitkina T, Gallo G and Spiliotis ET:
Septin-driven coordination of actin and microtubule remodeling
regulates the collateral branching of axons. Curr Biol.
22:1109–1115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mavrakis M, Azou-Gros Y, Tsai FC, Alvarado
J, Bertin A, Iv F, Kress A, Brasselet S, Koenderink GH and Lecuit
T: Septins promote F-actin ring formation by crosslinking actin
filaments into curved bundles. Nat Cell Biol. 16:322–334. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Agudelo-garcia PA, De Jesus JK, Williams
SP, Nowicki MO, Chiocca EA, Liyanarachchi S, Li PK, Lannutti JJ,
Johnson JK, Lawler SE, et al: Glioma cell migration on
three-dimensional nanofiber scaffolds is regulated by substrate
topography and abolished by inhibition of STAT3 signaling.
Neoplasia. 13:831–840. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rao JS: Molecular mechanisms of glioma
invasiveness: The role of proteases. Nat Rev Cancer. 3:489–501.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamaguchi H and Condeelis J: Regulation of
the actin cyto-skeleton in cancer cell migration and invasion.
Biochim Biophys Acta. 1773:642–652. 2007. View Article : Google Scholar
|
|
23
|
Sung BH, Zhu X, Kaverina I and Weaver AM:
Cortactin controls cell motility and lamellipodial dynamics by
regulating ECM secretion. Curr Biol. 21:1460–1469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yumura S, Itoh G, Kikuta Y, Kikuchi T,
Kitanishi-Yumura T and Tsujioka M: Cell-scale dynamic recycling and
cortical flow of the actin-myosin cytoskeleton for rapid cell
migration. Biol Open. 2:200–209. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Murrell MP and Gardel ML: F-actin buckling
coordinates contractility and severing in a biomimetic actomyosin
cortex. Proc Natl Acad Sci USA. 109:20820–20825. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vogel SK, Petrasek Z, Heinemann F and
Schwille P: Myosin motors fragment and compact membrane-bound actin
filaments. eLife. 2:e001162013. View Article : Google Scholar : PubMed/NCBI
|