Introduction
Intrahepatic cholangiocarcinoma (ICC) is an
epithelial cell malignancy arising from cholangiocytes within the
liver. Although ICC is rare, the morbidity and mortality rates have
markedly increased over the last three decades, particularly in
European and American countries (1). Unlike hepatic cellular carcinoma and
hilar cholangiocarcinoma, the dormant clinical symptoms and
ambiguous imaging features make it even harder for ICC to be
diagnosed at an early stage (2,3). Thus,
the chance for curative resection is generally limited, and
patients with ICC are also not suitable for liver transplantation
(4). Moreover, owing to its
desmoplastic character, complicated tumor microenvironment and rich
genetic heterogeneity, ICC is consistenly resistant to traditional
chemotherapeutics (5,6). Various targeted therapeutics and
combined chemotherapy do not demonstrate desired results except for
the protocol of gemcitabine plus cisplatin which achieves a median
survival time of only 11.7 months for patients who do not receive
surgery (7,8). Therefore, new sensitive therapeutics
are urgently needed.
Histone deacetylases (HDACs) are responsible for the
removal of acetyl groups from the lysine residues on the N-terminal
part of core histones (H2A, H2B, H3 and H4) and maintain a balance
with histone acetylase in normal organisms. When overexpressed,
HDACs may give rise to the formation of heterochromatin without DNA
binding by associated transcription factors. As a result, it may
silence various key genes, which play important roles in
transcriptional regulation, cell cycle progression and
developmental events. In the occurrence and development of ICC,
HDACs play an oncogene role (9).
Expression of HDAC1 was found to be significantly correlated with
lymph node metastasis, high stage carcinoma and vascular invasion
of ICC and was found to be negatively correlated with prognosis
(10). Therefore, it is reasonable
to regulate the expression and activity of HDACs for the treatment
of ICC. Although studies have reported the effects of various HDAC
inhibitors on bile duct cancer cells (11–13),
to the best of our knowledge, no research concerning the effects of
HDAC inhibitors on ICC has been reported to date.
HC toxin, a cyclic tetrapeptide first isolated from
the secondary metabolite of Helminthosporium carbonum
(14), is thought to be a type of
HDAC inhibitor in plants, insects and mammals (15). It was found to exhibit antitumor
activities in several types of human cancer cells with stronger
antitumor activity than other HDAC inhibitors in neuroblastoma
cells (16–18). In the present study, we explored the
effects of 34 types of HDAC inhibitors on ICC cell lines in
vitro, compared the anti-ICC activity of HC toxin with other
HDAC inhibitors and investigated the mechanisms involved in the
inhibitory effects of the HC toxin on ICC in vitro.
Materials and methods
Cell culture and HDAC inhibitors
ICC cell lines RBE and SSP-25 (obtained from Piken
University, Japan) and CCLP-1 and TFK-1 (obtained from the
University of Pittsburg, USA) were cultured in RPMI-1640 medium
(Gibco, USA) containing 10% fetal calf serum (FCS) (HyClone, USA).
All media contained 2 mM L-glutamine, 100 U/ml of penicillin G and
streptomycin. The cell lines were passaged twice/week and
maintained at 37°C in 5% CO2-95% air (vol/vol).
Trichostatin A (TSA), suberohydroxamic acid (SAHA), gemcitabine
(GEM), HC toxin and other HDAC inhibitors (Cayman 11076, no.
0466317) were dissolved in dimethyl sulfoxide (DMSO) at 10 mM ready
for usage.
Cell viability assay
Cells were plated in 96-well plates (at
5×103 cells/well), and divided into trial control and
blank groups co-cultured with HDAC inhibitors, solvent or nothing
for 48 h in three replicates, respectively. Cell viability was
verified by adding Cell Counting Kit-8 (CCK-8; Dojindo, Japan) into
the well at a final concentration of 10% after discarding the
previous supernatant and incubating for 2–4 h at 37°C. Then, the
absorbance at 450 nm was examined using a microplate reader
(Mithras LB 940; Berthold Technologies GmbH & Co., Bad Wildbad,
Germany) and displayed as the optical density (OD). Cell viability
= (trial group OD - blank group OD)/(control group OD - blank group
OD) × 100%.
Cell counting
Cells were plated in 6-well plates (at
5×104 cells/well). After culturing with the HDAC
inhibitors at different doses for 24–72 h with medium changed every
24 h, the cells were digested and dissolved using
phosphate-buffered saline (PBS) as single-cell suspension. Cell
number was examined using the Countess Cell Counting Chamber
(Invitrogen, USA).
Colony formation assay
Single-cell suspensions were plated into 6-well
plates at a density of 300–500 cells/well. After the cells became
adherent, the medium was replaced and different doses of HC toxin
were added. The cells were cultured for 10–14 days at 37°C and
stained with Giemsa staining after being fixed with 4%
paraformaldehyde (Sigma, USA). Viable colonies were counted. Colony
formation rate = colony number/number of plated cells × 100%.
Giemsa staining and light microscopic
observation
Cells were grown on coverslips placed in 6-well
plates (at 5×104/well) in advance and cultured with HC
toxin for two days. After being fixed with 4% paraformaldehyde and
washing with PBS three times/5 min, the cells were stained by
Giemsa staining (Xiangya, China) for 15–30 min and then placed on a
microscopic slide. An upright metallurgical microscope (Euro
immune; Germany) and an inverted microscope (Olympus, Japan) were
used to observe the morphology of the cells with and without Giemsa
staining, respectively. Images were captured and cells with
apoptotic bodies were counted.
Flow cytometry
The cells were harvest after incubation with the HC
toxin or solvent for 48 h. For apoptosis assessment, 7-AAD combined
with FITC-conjugated Annexin V (both from BioLegend, USA) were used
to treat the cells according to the manufacturer's protocols. For
cell cycle analysis, the cells were collected in 0.001% Triton of
PBS and stained with 7-AAD alone after being fixed in 5 ml of 70%
cold ethanol for 24 h. Approximately 10,000–20,000 cells were
analyzed with a FACSCalibur using CellQuest software (both from
Becton-Dickinson, USA). FlowJo 7.6.1 was used to analyze the
data.
Western blot analysis
Total proteins were extracted using the protein
extraction kit (KeyGen Biotech, China) and were transferred onto a
polyvinylidene fluoride membrane (Millipore, Germany) after being
separated on 10% SDS-PAGE. Anti-bax, anti-bcl-2, anti-cytochrome
c (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
anti-β-actin (Bioeasy, Korea), anti-caspase-3 (Abcam, USA),
anti-HDAC1 (Cell Signaling Technology, USA), and
anti-acetyl-histone H4 (Upstate) were used to detected proteins and
the homologous secondary antibody conjugated with horseradish
peroxidase were then used to detect the primary antibodies.
Immunoreactive bands were cast to X-ray film (Carestream, China)
after shining by a chemiluminescent substrate (Millipore, Germany).
ImageJ software (National Institutes of Health, Bethesda, MA, USA)
was used to calculate the relative expression of proteins.
Real-time PCR
Total RNA was extracted from the cells plated in
6-well plates after treatment with the HC toxin for 48 h using an
RNA simple extracted kit (Tiangen China). Reverse transcription
reaction was immediately performed using the PrimeScript RT reagent
kit (Takara, Japan). Real-Time PCR was carried out in an ABI 7500
Real-Time PCR system (Life Technologies) using the SYBR II-Green
PCR kit (Takara), and the data are shown in 2−ΔΔCt
format. β-actin was used as inner reference. Primers for HDAC1
were: 5′-CTCCATCCGTCCAGATAAAT-3′ and 5′-GCCACAGAACCACCAGTAG-3′; and
primers for β-actin were: 5′-GAGAAGGCGTGACATTAAG-3′ and
5′-GAAGGAAGGCTGGAAGAG-3′ (both from Sangon, China). All the
processes were performed according to the manufacturer's protocols
included in the kits, correspondingly.
Statistical analysis
Every experiment was performed at least three times
in triplicate, and data are shown as the mean ± SD. Differences
between groups were assessed using the Student's t-test (one sample
or paired t-tests), and were considered as statistically
significant at a P-value <0.05.
Results
Anti-ICC activity of HC toxin is superior
to that of other HDAC inhibitors and gemcitabine (GEM)
In the primary screening (Fig. 1A), all of the compounds at a
concentration of 3 µM were co-cultured with the REB cells
for 48 h and cell viability was assessed by the CCK-8 assay. The
results revealed that 13 of the 34 types of HDAC inhibitors showed
inhibitory activities with cell viability <100%. Among these,
the inhibitory effects of HC toxin, TSA, SAHA and 4-iodo-SAHA were
the most intensive with cell viability <60%.
To establish that the suppressive effects of the 13
HDAC inhibitors were not specific to the RBE cell line, we
co-cultured four ICC cell lines (TFK-1, RBE, SSP-25 and CCLP-1)
with three HDAC inhibitors (HC toxin, TSA or SAHA) or the positive
control (GEM) for 48 h. In the secondary screening (Fig. 1B), HC toxin was the most effective
followed by TSA, and all three HDAC inhibitors exhibited stronger
anti-ICC activities in the four ICC cell lines than GEM except for
SAHA in the SSP-25 cell line. The inhibitory activity of HC toxin
was similar to that of TSA in the TFK-1 and RBE cell lines while it
was superior to TSA in the SSP-25 and CCLP-1 cell lines from
concentration from ~30 nM to 8 µM. These results indicated
that the anti-ICC activity of HC toxin was superior to that of the
other HDAC inhibitors and GEM.
We also calculated the IC50 values of the
HC toxin. For the CCLP-1, SSP-25, TFK-1 and RBE cell lines, the
IC50 values were ~297.6±80.4, 520.0±43.0, 854.6±86.9 and
713.7±27.3 nM, respectively. Thus, we chose HC toxin at a
concentration ~400 nM to co-culture with the CCLP-1 cells in the
following experiments.
HC toxin suppresses cell proliferation
and colony formation of CCLP-1 cells
HC toxin (0–400 nM) was co-cultured with CCLP-1
cells, and cell number decreased with increasing concentration of
HC toxin for 24–72 h (Fig. 2A). HC
toxin at 100 nM did not significantly restrict the cell
proliferation of the CCLP-1 cells at the three time points. At 48
h, the number of cells was reduced by 1.5- to 2.2-fold following
treatment with HC toxin in a dose-dependent manner which was in
line with the cell viability assay as determined by the CCK-8
assay.
In the colony formation assay (Fig. 2B and C), the colony formation rate
of the CCLP-1 cells was decreased from ~53.1±5.2% in the control
group (0 nM) to 0% in the 400 nM group. The rate of colony
formation of the 50 nM group was modestly increased when compared
with the 0 nM group, but the difference was not statistically
significant.
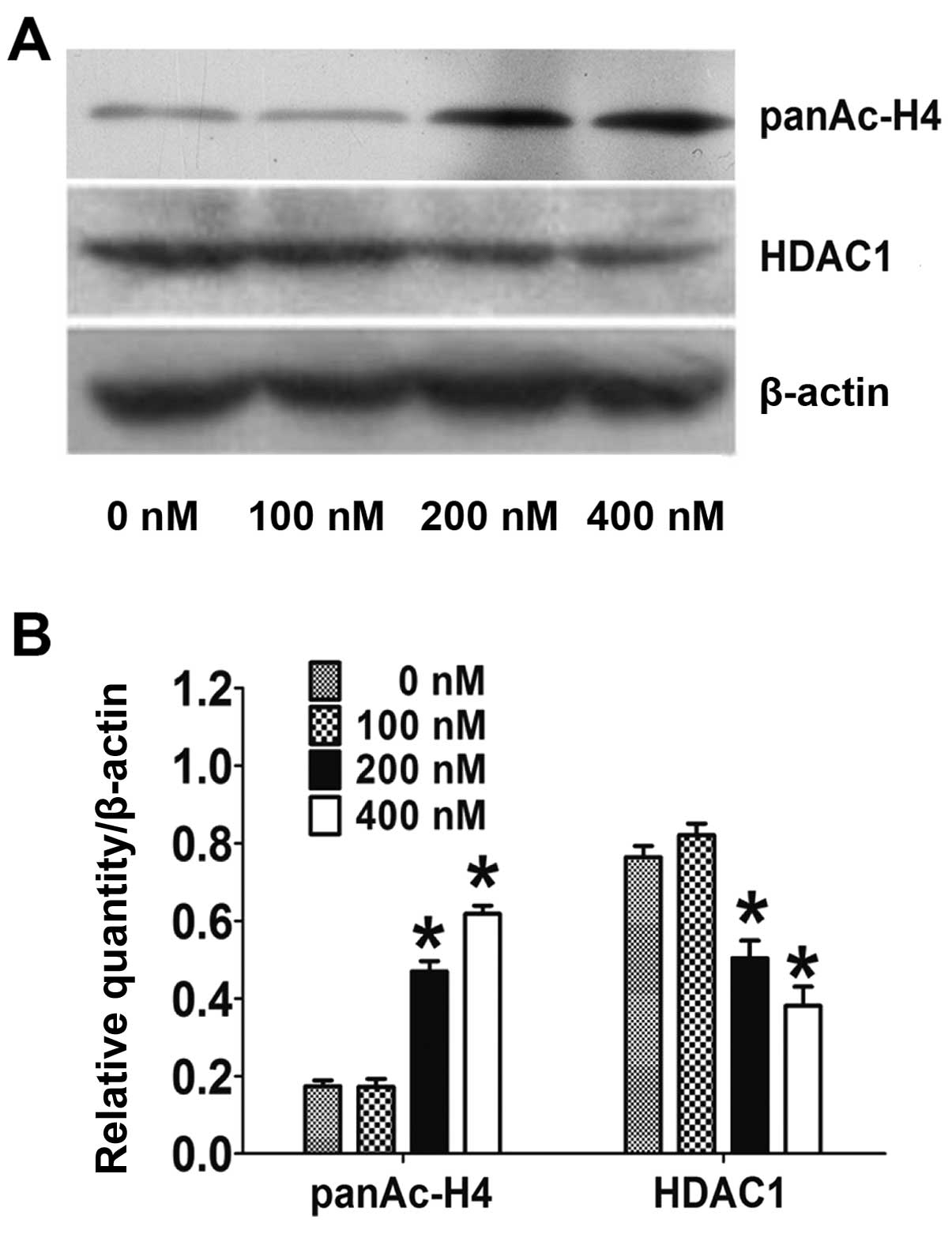
HC toxin downregulates the protein level
of acetyl-histone H4 and HDAC1
The activity of HDAC was examined indirectly by
checking the level of acetyl-histone H4. As shown in Fig. 3A and B, the acetyl-histone H4 level
was markedly reduced by 2.5- to 3.3-fold following treatment with
200 and 400 nM of HC toxin, respectively, while the level did not
obviously change following HC toxin treatment at 100 nM. This
correlated with the inhibitory effects on the cell proliferation
and colony ability in the CCLP-1 cells.
We also examined the expression of HDAC1 in CCLP-1
cells. As shown in Fig. 3A and B,
the expression of HDAC1 protein was markedly reduced 1.6- to
2.2-fold at 200 and 400 nM of HC toxin respectively while no change
was obvious at 100 nM. However, the mRNA of HDAC1 examined by
real-time PCR did not change at all of the three doses used
(Fig. 3C).
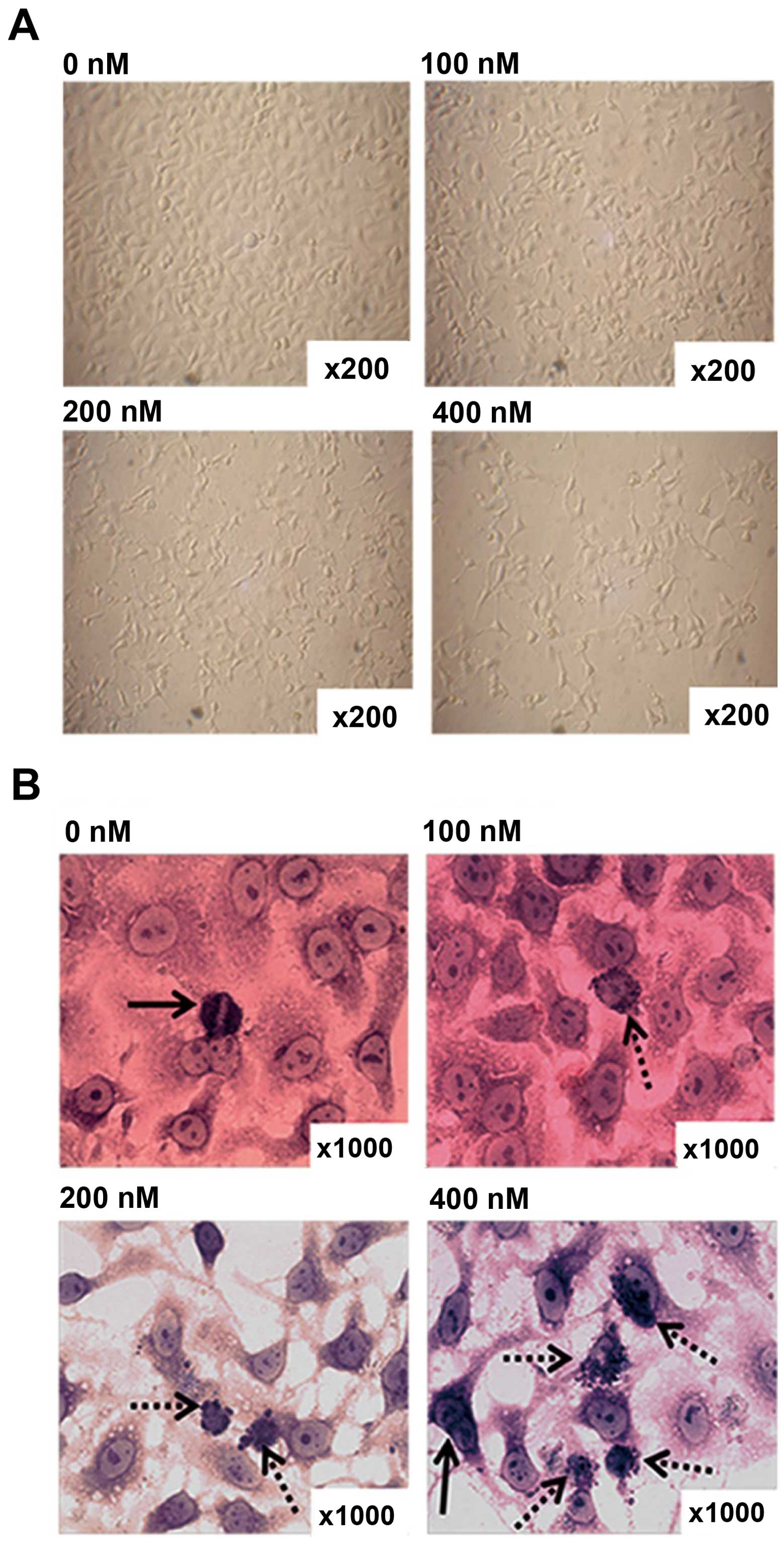
HC toxin induces multiple morphological
changes under light microscopic observation
CCLP-1 cells were cultured with the different doses
of HC toxin for 48 h. The morphology and growth features of the
cells in each groups were observed by light microscopy before and
after Giemsa staining. With increasing doses of HC toxin, the cells
gradually decreased in density with single cells dwindling in size,
dendrite-like structures appearing and gradually became longer
(Fig. 4A).
In order to clearly observe cell nuclei, Giemsa
staining was performed. As shown in Fig. 4B, multinucleated and cellular atypia
were reduced, mitotic figures decreased, apoptotic bodies appeared
and progressively increased with the increasing concentration of
the HC toxin. We calculated the number of cells with apoptotic
bodies from 10 fields with a magnification of x200. This showed
that the number of cells with apoptotic bodies in the 200 and 400
nM groups was much higher than that in the 0 and 100 nM groups, and
there was no difference between the latter two in terms of
statistical significance (Fig.
4C).
Cell cycle is arrested at the G0/G1
stage
Cell cycle distribution was detected by flow
cytometry with 7-AAD staining. After incubation with HC toxin for
48 h, the floating cells were removed to exclude the interference
from apoptosis and dead cells. Compared with the 0 nM group, the 20
and 400 nM groups exhibited significantly higher percentages of
cells in the G0/G1 stage and the percentages of cells in the G2/M
stage were markedly deceased in a dose-dependent manner (Fig. 5A and B). No difference in the
percentage of cells in the S stage was observed among the groups.
This illustrated that HC toxin arrests the cell cycle in the G0/G1
stage.
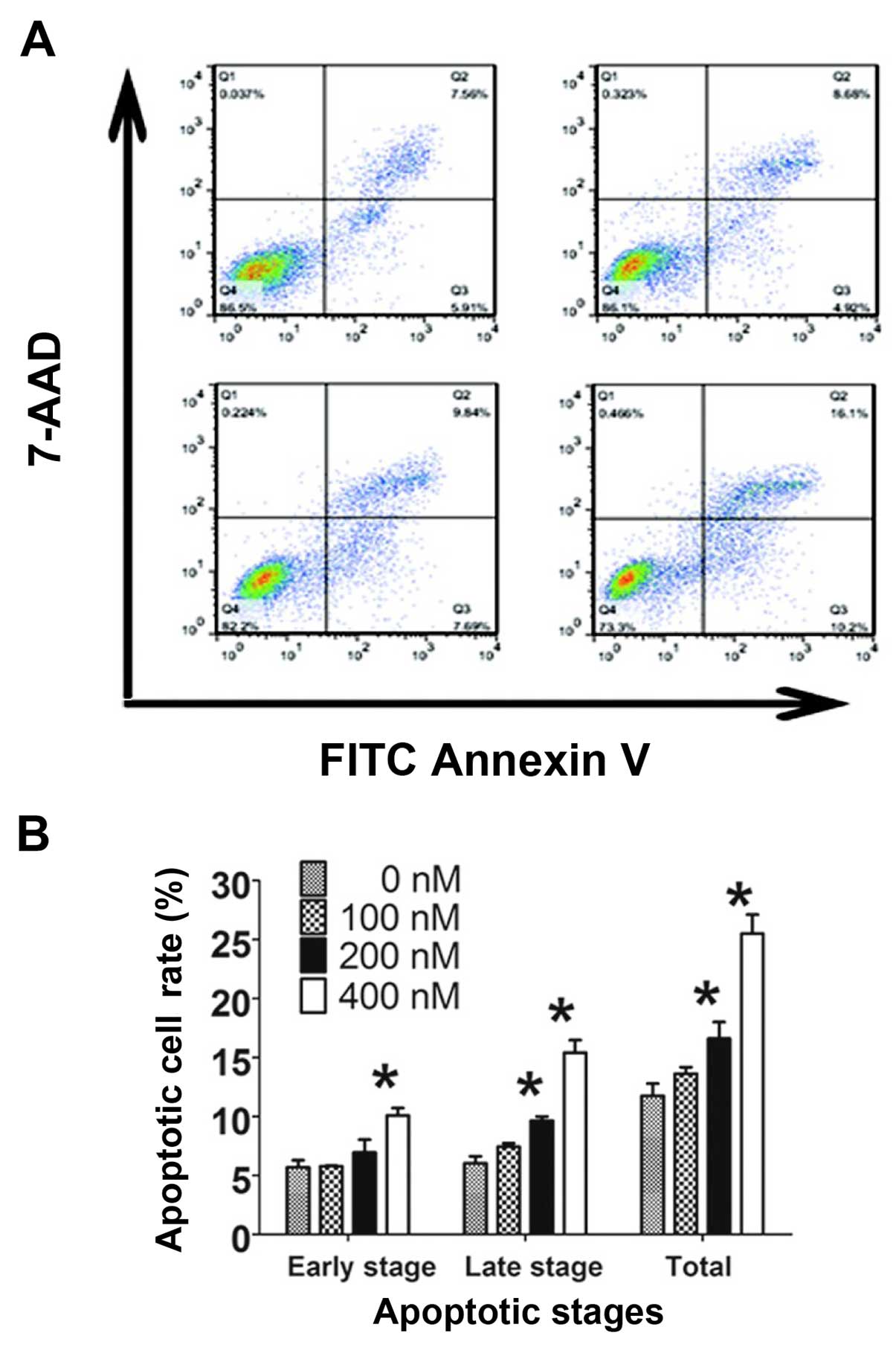
HC toxin induces cell apoptosis via the
caspase-3-dependent and -independent pathways
The apoptotic effects of the HC toxin were examined
by flow cytometry with staining of Annexin V-FITC combined with
7-AAD and by assessment of morphological changes with Giemsa
staining. In the 200 and 400 nM groups, the numbers of cells with
apoptotic bodies were significantly increased compared with the 0
nM group (Fig. 4C). The percentages
of total apoptosis, early apoptosis and late apoptosis were
increased with the increasing HC toxin concentration (Fig. 6A and B). Significant increases in
the percentages of cells in the late and total stage of apoptosis
were noted at 200 nM of HC toxin, but could not be observed in the
early stage. This may be due to the binding capacity of Annexin
V-FITC or the instrument noise.
We then explored the specific apoptosis pathways
involved by assessing the expression changes in apoptotic proteins.
The expression of caspase-3 did not show a significant increase
until the concentration of HC toxin increased to 400 nM with only
an increase of ~1.5-fold. The levels of bax/bcl-2 and cytochrome
c did not change in our experimental environment (Fig. 6C and D). Therefore, it is reasonable
to consider that the caspase-3-dependent pathway was not the major
pathway which was involved in the HC toxin-mediated apoptosis while
the caspase-3-independent pathway may play the major role.
Discussion
ICC is the second most common malignant tumor in the
liver and its pathogenesis is still unclear. Late diagnosis and the
high recurrence rate have limited the effects of surgical excision
and lead to dismal overall 5- and 3-year survival rates even worse
than HCC (19). Moreover, there is
no effective chemotherapy regimens for ICC due to the high
chemoresistance. Thus, discovery of novel sensitive therapeutics is
warranted. In the present study, we carried out two rounds of
screening for 34 types of HDAC inhibitors. HC toxin was the most
effective in the suppression of growth of the ICC cell lines and
the mechanisms were associated with cell apoptosis, cell cycle
arrest and differentiation.
HDAC inhibitors are potential antitumor agents
composed of hydroxamic acid, cyclotetrapeptides, benzamide, some
short chain fatty acids and other molecular structure types
(20). The 34 HDAC inhibitors used,
which were chosen from an epigenetic library provided by Cayman
Co., covered more molecular structure types of HDAC inhibitors than
the previous study (16). In the
primary screening, 13 HDAC inhibitors exhibited anti-ICC activity
and four exhibited intensive activity (cell viability <60%).
Among them, valproic acid (VPA), which was the earliest HDAC
inhibitor, exhibited a weak antitumor activity in RBE cells in line
with previous research on HuCCT1 (ICC cell line) and SUIT-2
(pancreas cancer cell line) cells (13). Yet, the significance of the primary
screening was confined to only one cell line and one concentration
was used. Therefore, we performed a secondary screening in four ICC
cell lines with different doses of HDAC inhibitors. In the
secondary screening, HC toxin showed an anti-ICC activity much
stronger than that of gemcitabine which is currently one of the
most important chemotherapeutic for the treatment of ICC (21,22).
The effect of TSA was similar to that of HC toxin in RBE and TFK-1
cells but was obviously inferior to the latter in SSP-25 and CCLP-1
cells. This suggested that HC toxin may have a wider antitumor
spectrum and may also work in the treatment of some tumor subtypes
with chemoresistance. Hence, HC toxin could be a potential
therapeutic for cancer treatment.
The mechanisms involved in the inhibitory effects of
the HC toxin in ICC cell lines are various and are as follows.
Firstly, as a cell-permeable, reversible inhibitor
of HDACs, HC toxin acts mainly by inhibiting the activities of
HDACs to carry out H3 and/or H4 hyperacetylation in their promoter
regions (15) and this is in line
with the results shown in Fig. 3A and
B. In our research, we also found that the protein expression
of HDAC1 decreased by western blot analysis. However, the mRNA of
HDAC1 did not change correspondingly. This illustrated that HC
toxin downregulated the HDAC1 level depending on the
post-transcriptional modification or the degradation acceleration
of HDAC1 itself.
Secondly, the cell cycle was arrested at the G0/G1
stage which suggested that DNA replication or histone synthesis was
blocked at the S stage. The results were in line with previous
findings of HC toxin in neuroblastoma cells (16), but not breast cancer cells (T47D) in
which the cell cycle was arrested at G2/M (17). This difference may be associated
with the nature of the cell lines used. Cell cycle arrest at G0/G1
may rely on a change in p21waf1 and cyclin protein
(23). As a cell cyclin kinase
inhibitor, p21waf1 upregulation may suppress the
activities of cyclin and reduce the phosphorylation level of Rb
therefore resulting in the decrease in the E2F level and finally
arresting the cell cycle at the G0/G1 stage. Suberoylanilide
hydroxamic acid (SAHA), another type of HDAC inhibitor, induces a
sharp rising in p21waf1 protein and mRNA of bladder
carcinoma cells (T24) (24). Many
other HDAC inhibitors have a similar functional mechanism (25–27),
and this may also exist in the process of HC toxin function in
CCLP-1 cells; yet this requires further experiments to verify.
Additionally, HC toxin may play a role in cell cycle
synchronization and may be a solution for drug-resistance in some
cases; thus, the use of HC toxin in various combination
chemotherapy protocols should be considered.
Thirdly, morphologic changes and flow cytometry
demonstrated that HC toxin strongly induced the apoptosis of CCLP-1
cells. Cell apoptotic pathways can be simply divided into
caspase-3-dependent and caspase-3-independent pathways (28). Bax/Bcl-2, cytochrome c and
caspase-3 are the major apoptosis-related proteins in the former
containing endogenous and exogenous pathways (29,30).
In the caspase-3-independent pathway, AIF, Endo G are the primary
functional proteins produced in mitochondria and transferred into
the nucleus to cleave DNA directly (31). In the western blot assay, the
caspase-3 level was not markedly increased until the concentration
of the HC toxin increased to 400 nM which did not correlate with
the results of the flow cytometry. In addition, the Bax/Bcl-2 ratio
and the level of cytochrome c did not significantly change
at all HC toxin doses used. In a previous study performed in breast
cancer cells, HC toxin did not obviously change the level of
caspase-3 as well (17). This
demonstrated that HC toxin induced apoptosis by means of both the
caspase-3-dependent and -independent pathways and the latter could
be dominant.
Finally, it is likely that HC toxin may regulate the
differentiation of CCLP-1 cells according to the morphologic
changes consisting of the appearance of dendritic-like structures
(32). In addition, it was
confirmed that HC toxin promotes the differentiation of
neuroendocrine cells (33).
However, this finding should be further investigated at a molecular
level to provide further evidence in order to establish whether or
not HC toxin induces the differentiation of ICC cell lines.
In summary, HC toxin demonstrated anti-ICC activity
superior to that of other HDAC inhibitors and GEM. It suppressed
cell proliferation, arrested the cell cycle and induced apoptosis
and may induce the differentiation of ICC cells in vitro.
These findings provide the experimental basis for the treatment of
ICC by HC toxin.
Acknowledgments
The present study was supported by grants from the
Natural Science Foundation of China (no. 81071990), the Guangdong
Province Natural Science Foundation (no. 2015A030313725), and the
Guangdong Science Province and Technology Program projects
(2012B03180411).
References
|
1
|
Everhart JE and Ruhl CE: Burden of
digestive diseases in the United States Part III: Liver, biliary
tract, and pancreas. Gastroenterology. 136:1134–1144. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iavarone M, Piscaglia F, Vavassori S,
Galassi M, Sangiovanni A, Venerandi L, Forzenigo LV, Golfieri R,
Bolondi L and Colombo M: Contrast enhanced CT-scan to diagnose
intrahepatic cholangiocarcinoma in patients with cirrhosis. J
Hepatol. 58:1188–1193. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim SH, Lee CH, Kim BH, Kim WB, Yeom SK,
Kim KA and Park CM: Typical and atypical imaging findings of
intrahepatic cholangiocarcinoma using gadolinium ethoxybenzyl
diethylenetriamine pentaacetic acid-enhanced magnetic resonance
imaging. J Comput Assist Tomogr. 36:704–709. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Robles R, Figueras J, Turrión VS, Margarit
C, Moya A, Varo E, Calleja J, Valdivieso A, Valdecasas JC, López P,
et al: Spanish experience in liver transplantation for hilar and
peripheral cholangiocarcinoma. Ann Surg. 239:265–271. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tepsiri N, Chaturat L, Sripa B, Namwat W,
Wongkham S, Bhudhisawasdi V and Tassaneeyakul W: Drug sensitivity
and drug resistance profiles of human intrahepatic
cholangiocarcinoma cell lines. World J Gastroenterol. 11:2748–2753.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kiefer MV, Albert M, McNally M, Robertson
M, Sun W, Fraker D, Olthoff K, Christians K, Pappas S, Rilling W,
et al: Chemoembolization of intrahepatic cholangiocarcinoma with
cisplatinum, doxorubicin, mitomycin C, ethiodol, and polyvinyl
alcohol: A 2-center study. Cancer. 117:1498–1505. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee J, Park SH, Chang HM, Kim JS, Choi HJ,
Lee MA, Jang JS, Jeung HC, Kang JH, Lee HW, et al: Gemcitabine and
oxaliplatin with or without erlotinib in advanced biliary-tract
cancer: A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 13:181–188. 2012. View Article : Google Scholar
|
|
8
|
Valle J, Wasan H, Palmer DH, Cunningham D,
Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira
SP, et al: ABC-02 Trial Investigators: Cisplatin plus gemcitabine
versus gemcitabine for biliary tract cancer. N Engl J Med.
362:1273–1281. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sriraksa R and Limpaiboon T: Histone
deacetylases and their inhibitors as potential therapeutic drugs
for cholangiocarcinoma - cell line findings. Asian Pac J Cancer
Prev. 14:2503–2508. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morine Y, Shimada M, Iwahashi S,
Utsunomiya T, Imura S, Ikemoto T, Mori H, Hanaoka J and Miyake H:
Role of histone deacetylase expression in intrahepatic
cholangiocarcinoma. Surgery. 151:412–419. 2012. View Article : Google Scholar
|
|
11
|
Zhang P, Guo Z, Wu Y, Hu R, Du J, He X,
Jiao X and Zhu X: Histone deacetylase inhibitors inhibit the
proliferation of gallbladder carcinoma cells by suppressing
AKT/mTOR signaling. PLoS One. 10:e1361932015.
|
|
12
|
Kitamura T, Connolly K, Ruffino L, Ajiki
T, Lueckgen A, DiGiovanni J and Kiguchi K: The therapeutic effect
of histone deacetylase inhibitor PCI-24781 on gallbladder carcinoma
in BK5.erbB2 mice. J Hepatol. 57:84–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iwahashi S, Ishibashi H, Utsunomiya T,
Morine Y, Ochir TL, Hanaoka J, Mori H, Ikemoto T, Imura S and
Shimada M: Effect of histone deacetylase inhibitor in combination
with 5-fluorouracil on pancreas cancer and cholangiocarcinoma cell
lines. J Med Invest. 58:106–109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Islam MN, Islam MS, Hoque MA, Kato T,
Nishino N, Ito A and Yoshida M: Bicyclic tetrapeptides as potent
HDAC inhibitors: Effect of aliphatic loop position and
hydrophobicity on inhibitory activity. Bioorg Med Chem.
22:3862–3870. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Walton JD: HC-toxin. Phytochemistry.
67:1406–1413. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deubzer HE, Ehemann V, Kulozik AE,
Westermann F, Savelyeva L, Kopp-Schneider A, Riester D, Schwab M
and Witt O: Anti-neuroblastoma activity of Helminthosporium
carbonum (HC)-toxin is superior to that of other differentiating
compounds in vitro. Cancer Lett. 264:21–28. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Joung KE, Kim DK and Sheen YY:
Antiproliferative effect of trichostatin A and HC-toxin in T47D
human breast cancer cells. Arch Pharm Res. 27:640–645. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kamitani H, Taniura S, Ikawa H, Watanabe
T, Kelavkar UP and Eling TE: Expression of 15-lipoxygenase-1 is
regulated by histone acetylation in human colorectal carcinoma.
Carcinogenesis. 22:187–191. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Khan SA, Davidson BR, Goldin RD, Heaton N,
Karani J, Pereira SP, Rosenberg WM, Tait P, Taylor-Robinson SD,
Thillainayagam AV, et al: British Society of Gastroenterology:
Guidelines for the diagnosis and treatment of cholangiocarcinoma.
An update Gut. 61:1657–1669. 2012. View Article : Google Scholar
|
|
20
|
Jung M: Inhibitors of histone deacetylase
as new anticancer agents. Curr Med Chem. 8:1505–1511. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weber SM, Ribero D, O'Reilly EM, Kokudo N,
Miyazaki M and Pawlik TM: Intrahepatic cholangiocarcinoma: Expert
consensus statement. HPB. 17:669–680. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bridgewater J, Galle PR, Khan SA, Llovet
JM, Park JW, Patel T, Pawlik TM and Gores GJ: Guidelines for the
diagnosis and management of intrahepatic cholangiocarcinoma. J
Hepatol. 60:1268–1289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Richon VM, Sandhoff TW, Rifkind RA and
Marks PA: Histone deacetylase inhibitor selectively induces
p21WAF1 expression and gene-associated histone
acetylation. Proc Natl Acad Sci USA. 97:10014–10019. 2000.
View Article : Google Scholar
|
|
24
|
Yamaguchi J, Sasaki M, Sato Y, Itatsu K,
Harada K, Zen Y, Ikeda H, Nimura Y, Nagino M and Nakanuma Y:
Histone deacetylase inhibitor (SAHA) and repression of EZH2
synergistically inhibit proliferation of gallbladder carcinoma.
Cancer Sci. 101:355–362. 2010. View Article : Google Scholar
|
|
25
|
Sachweh MC, Drummond CJ, Higgins M,
Campbell J and Laín S: Incompatible effects of p53 and HDAC
inhibition on p21 expression and cell cycle progression. Cell Death
Dis. 4:e5332013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Emori T, Kitamura K and Okazaki K: Nuclear
Smad7 overexpressed in mesenchymal cells acts as a transcriptional
corepressor by interacting with HDAC-1 and E2F to regulate cell
cycle. Biol Open. 1:247–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li L, Dai HJ, Ye M, Wang SL, Xiao XJ,
Zheng J, Chen HY, Luo YH and Liu J: Lycorine induces cell-cycle
arrest in the G0/G1 phase in K562 cells via HDAC inhibition. Cancer
Cell Int. 12(49)2012. View Article : Google Scholar
|
|
28
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Laulier C and Lopez BS: The secret life of
Bcl-2: Apoptosis-independent inhibition of DNA repair by Bcl-2
family members. Mutat Res. 751:247–257. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Candé C, Cohen I, Daugas E, Ravagnan L,
Larochette N, Zamzami N and Kroemer G: Apoptosis-inducing factor
(AIF): A novel caspase-independent death effector released from
mitochondria. Biochimie. 84:215–222. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Scott RE: Differentiation,
differentiation/gene therapy and cancer. Pharmacol Ther. 73:51–65.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Deubzer HE, Ehemann V, Westermann F,
Heinrich R, Mechtersheimer G, Kulozik AE, Schwab M and Witt O:
Histone deacetylase inhibitor Helminthosporium carbonum (HC)-toxin
suppresses the malignant phenotype of neuroblastoma cells. Int J
Cancer. 122:1891–1900. 2008. View Article : Google Scholar
|