Introduction
Chronic myeloid leukemia (CML) is a type of
hematological malignancy caused by the neoplastic transformation of
hematopoietic stem cells (1). CML
is characterized by a reciprocal chromosomal translocation
t(9;22)(q34;q11) named the Philadelphia chromosome which fuses the
Abelson kinase (ABL) gene from chromosome 9 with the breakpoint
cluster region (BCR) gene on chromosome 22 (2). The BCR-ABL fusion oncogene is
responsible for the pathogenesis of CML and is the primary
molecular target for therapy with imatinib mesylate (formerly
ST1571), a tyrosine kinase inhibitor (3,4).
Although imatinib treatment has achieved outstanding curative
efficacy in the chronic phase of CML, a significant portion of
patients fail drug treatment due to intrinsic or acquired drug
resistance (5). The drug resistance
mechanisms involve many factors, which are mainly divided into
BCR-ABL-dependent and BCR-ABL-independent mechanisms (6). Among the latter is the MDR phenotype
which is dependent on the expression of drug resistance proteins
represented by P-gp (7). P-gp is
encoded by the MDR1 gene and belongs to the superfamily of
ATP-binding cassette transporters, which powerfully pump a wide
variety of structurally diverse chemotherapeutics across the
membranes, resulting in a decreased intracellular drug
concentration and chemotherapy bioavailability (8). It has been demonstrated that imatinib
is a substrate of the P-gp pump (9). Consequently, much interest is focused
on searching for drug transport antagonists for these proteins or
identifying proliferation inhibitors of MDR cancer cells.
There are various PI3K families found in higher
eukaryotes. To date, the class IA PI3Ks consisting of a p110
catalytic subunit and a p85 regulatory subunit have been implicated
in cancer (10). Multiple growth
factors such as IGF, PDGF, EGF and HGF can activate PI3K (11–14).
Following PI3K activation, phosphoinositol-4,5-bisphosphate (PIP2)
is converted into the second messenger
phosphoinositol-3,4,5-trisphosphate (PIP3) (15). The serine-threonine kinase Akt
containing the pleckstrin homology domain binds to PIP3 at the
plasma membrane where Akt undergoes phosphorylation at both Thr308
and Ser473 (16). The aberrant
activation of the PI3K/Akt cell transduction pathway has been found
in different types of neoplasms, which contributes collectively to
inhibit cell apoptosis and promote tumor proliferation,
angiogenesis, invasion and metastasis (17). Recently, involvement of the PI3K/Akt
signaling pathway in MDR has attracted increased attention.
Research has highlighted that the PI3K/Akt pathway is closely
associated with modulation of MDR mediated by P-gp in human
leukemia, breast carcinoma, and gastric cancer (18–20).
Osthole, 7-methoxy-8-(3-methyl-2-butenyl) coumarin,
is extracted from the fruit of Cnidium monnieri (L.) Cusson
and is widely used in traditional Chinese medicine. Osthole
exhibits a broad spectrum of pharmacological and biological
properties such as anti-hepatitis, anti-osteoporosis,
anti-inflammatory, anti-allergic and anti-mutation (21). Moreover, several publications have
indicated that osthole possesses neuroprotective effects against
traumatic brain injury and accumulation of β-amyloid peptide
injuries (22). Osthole has
recently been confirmed to exhibit effective anticancer activity
either in vivo or in vitro. Nevertheless, comparative
experiments suggest that osthole causes neither apoptosis nor
growth inhibiting effects on normal human peripheral blood
mononuclear cells and cervical cells (23). Accumulating evidence has revealed
that osthole can suppress proliferation as well as enhance
apoptosis, invasion and migration in human lung cancer, breast
carcinoma, hepatocellular carcinoma, cervical cancer, colorectal
adenocarcinoma, and glioblastoma multiforme (24–29).
But to the best of our knowledge, little research has been
conducted to explore the function of osthole in hematopoietic
malignancies, which was only reported in the human acute
promyelocytic leukemia HL-60 cell line (24). On the other hand, the efficacy and
the associated molecular mechanisms of osthole in the reversal of
MDR, to date, have not been examined in MDR leukemia cells.
In the present study, we utilized the human
myelogenous leukemia K562/ADM cell line which is resistant to
doxorubicin chemotherapy and has been extensively applied for
academic studies. We investigated the intrinsic cytotoxic effect of
osthole on K562/ADM cells. The reversal ability of osthole and the
potential linkage of the reversal of P-gp-mediated MDR and the
PI3K/Akt signaling cascade were described, which may be the major
mechanisms underlying the MDR reversal potential of osthole in
K562/ADM cells.
Materials and methods
Chemicals and reagents
Osthole was obtained from the National Institute for
the Control of Pharmaceutical and Biological Products (Beijing,
China). Osthole was dissolved in dimethyl sulfoxide (DMSO;
Sigma-Aldrich, St. Louis, MO, USA) as a stock solution (10 mM),
stored at −20°C, and diluted in RPMI-1640 medium (Hyclone
Laboratories, Inc., Logan, UT, USA) to the desired concentration
before each experiment. The final DMSO concentration did not exceed
0.1% throughout the study. Doxorubicin (Melone Pharmaceutical Co.,
Ltd., Dalian, China) was dissolved in a stock concentration of 10
mM with ddH2O and divided into five aliquots. Monoclonal
rabbit anti-human MDR1 (cat no. 12683), poly-clonal rabbit
anti-human AKT (cat no. 9272) and polyclonal rabbit anti-human
phospho-AKT (Ser473, cat no. 9271) were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Polyclonal rabbit
anti-human GAPDH (cat no. bs-2188R) was a product of Biosynthesis
Biotechnology Co., Ltd. (Beijing, China).
Cell lines and cell culture
The parental drug-sensitive human CML K562 cell line
was supplied by the Key Laboratory of Tumour Molecular Biology of
Binzhou Medical University (Binzhou, China) and the MDR K562/ADM
subline was obtained from the Department of Pharmacology, The
Institute of Hematology of the Chinese Academy of Medical Sciences
(Tianjin, China). The two cell lines were maintained in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS; Hyclone), in
a humidified atmosphere of 5% CO2 at 37°C. The K562/ADM
cells were grown in the presence of 1 µM doxorubicin. Before
the experiment, doxorubicin was withdrawn from the cells for two
weeks.
Determination of multidrug
resistance
To determine the multidrug resistance of the
K562/ADM cells to chemotherapeutic agents, K562/ADM cells and the
parental K562 cells (1×104/well) were seeded into
96-well plates and incubated with multiple concentrations of
doxorubicin, diluted in RPMI-1640, for 24 h. Each group consisted
of five parallel wells. After that, 10 µl of
2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium
monosodium salt (WST-8) (Cell Counting Kit-8, CCK-8; Dojindo
Molecular Technologies, Inc., Shanghai, China) solution was added
to every well and incubated for a further 4 h. Then, the absorbance
at 570 nm was measured using a fluorescence spectrofluorometer
(F-7000; Hitachi High-Technologies Corporation, Tokyo, Japan).
After plotting the dose-effect curve, IC50 values and
the resistant fold obtained from the data [Resistant fold =
IC50 of K562/ADM cells/IC50 of K562 cells]
were calculated. Each experiment was carried out in triplicate.
Intrinsic cytotoxicity assay
The cytotoxicity of osthole in vitro was
assessed using the CCK-8 assay. In short, K562/ADM cells at the
logarithmic growth phase were seeded into a 96-well plate at a
density of 1×104 cells/well. Subsequently, osthole
diluted in RPMI-1640 was added. The cells were cultured in a
humidified incubator in 5% CO2 at 37°C for 24 h, and the
quantity of viable cells was determined. Because the concentrations
of the reversal multidrug agents we needed were neither inhibitory
nor toxic (30), osthole
concentrations of 5 and 15 µM were finally used to study the
reversal of MDR. All analyses were carried out at least thrice
independently.
Assay of the reversal efficacy
The potential of osthole to overcome MDR was
evaluated by the CCK-8 method in the K562/ADM cell line. K562/ADM
cells were seeded into 96-well plates (~1×104 cells each
well). Shortly afterward, doxorubicin only or combinations with
osthole (5 and 15 µM) diluted in RPMI-1640 were added. The
cells were maintained at 37°C in a humidified atmosphere of 5%
CO2 for 24 h, and the percentages of viable cells were
appraised by the CCK-8 assay. Each group included five parallel
wells. The reversal fold (RF) values were calculated using the
following formula: RF = IC50 of doxorubicin alone/the
IC50 of doxorubicin in the presence of osthole.
Enhancement of Rhodamine-123 (Rh-123)
accumulation
ABC transporters especially P-gp play essential
roles in the efflux of chemotherapeutic drugs from tumor cells into
the surrounding tissue (8). To
detect the capacity of osthole on the P-gp pump, K562/ADM or K562
cells were pretreated in the absence or presence of osthole at
concentrations of 5 and 15 µM in an incubator at 37°C and 5%
CO2 in air for 24 h. Then, Rh-123 at a final
concentration of 5 µM was applied to the wells. After
incubation for 30 min, the cells were harvested by centrifugation
and washed thrice with ice-cold PBS. The intracellular mean
fluorescence intensity (MFI) of Rh123 was measured using a flow
cytometer (FACS FC500; Beckman Coulter, Brea, CA, USA) with an
excitation wavelength of 488 nm and emission wavelength of 530
nm.
Enhancement of intracellular doxorubicin
accumulation
A standard procedure was employed to monitor the
intracellular accumulation of doxorubicin. Briefly, K562/ADM or
K562 cells (5×105/well) were seeded into 6-well plates.
The cells were pretreated in the absence or presence of osthole at
concentrations of 5 and 15 µM in the dark at 37°C and 5%
CO2 in air for 24 h. Then, doxorubicin (5 µM) was
applied to the wells. After incubation for 1 h, the cells were
collected and washed three times with ice-cold PBS. The
intracellular MFI associated with doxorubicin was detected by FACS.
Excitation wavelength and emission wavelength were 485 and 580 nm,
separately.
RNA isolation and reverse
transcription-PCR (RT-PCR) analysis
Previous to PCR, K562/ADM cells were pretreated with
osthole (5, 15 µM) or RPMI-1640 for 24 h. Total RNA was
isolated using TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA) in accordance with the manufacturer's protocol.
The purity of the total RNA was assessed from the ratio of
A260/A280 by a spectrophotometer (NanoDrop 2000). RNA (1–2
µg) was applied for the synthesis of the first strand cDNA.
The primers used in this experiments were designed using Primer 5
version 5.6.0 software and synthesized by Sangon Biotech Co., Ltd.
(Shanghai, China). The primers and probe sets were as follows: MDR1
forward, 5′-GTTGCCATTGACTGAAAGAAAGAAC-3′ and reverse,
5′-ACAGGAGATAGGCTGGTTTGA-3′; 126 bp); and GAPDH forward,
5′-TGACTTCAACAGCGACACCCA-3′ and reverse,
5′-CACCCTGTTGCTGTAGCCAA-3′; 121 bp). The reverse transcription
reaction was implemented with PrimeScript™ RT reagent kit with gDNA
Eraser (Takara Bio, Otsu, Shiga, Japan). The polymerase chain
reaction amplification was performed on Eppendorf Mastercycler
Personal (Eppendorf China Co., Ltd.) by using Premix Taq™ (Takara).
The reaction system contained diethyl pyrocarbonate, forward
primer, reverse primer, Premix Taq and template cDNA. After 5 min
of initial incubation at 95°C, cDNA was amplified in 35 cycles
consisting of a 40-sec denaturation at 95°C, a 30-sec annealing at
56°C and a 40-sec elongation at 72°C. The PCR products were
separated by 1.5% agarose gels (Takara Biotechnology Dalian Co.,
Ltd.), and stained with ethidium bromide for 15 min. The images
were captured by Tanon GIS system. GAPDH served as an internal
standard for quality control and quantification of the target
genes.
Western blot assay
K562 and K562/ADM cells following different
treatments were harvested, and the proper amount of lysis buffer
(Beyotime Biotechnology, Shanghai, China) was added. Protein
concentrations were determined by a bicincho-ninic acid protein
assay kit (Beyotime Biotechnology). Protein samples were separated
on 10 or 6% SDS-PAGE gel (Beyotime Biotechnology) and transferred
onto polyvinylidine difluoride (PVDF) membranes (EMD Millipore,
Bedford, MA, USA). The membranes were blocked with 5% skimmed dry
milk in TBS containing 0.2% Tween-20 at room temperature for 2 h
and incubated overnight at 4°C with primary antibodies against MDR1
(1:1,000), AKT (1:1,000), p-Akt (Ser473, 1:1,000) and GADPH
(1:500). After three washes in TBS/Tween buffer, the membranes were
incubated in horesradish peroxidase-labeled goat anti-rabbit
immunoglobulin G (1:5,000; cat no. ZB-5301; Beijing
ZhongShan-Golden Bridge Technology Co., Ltd., Beijing, China) for 2
h at 37°C. Detection was performed using the FluorChem FC2 gel
imaging system (Alpha Innotech, San Leandro, CA, USA). Each band
density was quantified using ImageJ image processing program and
normalized by GAPDH for their respective lanes.
Data analysis
Statistical analyses were carried out using SPSS13.0
software (IBM SPSS, Armonk, NY, USA). Data are expressed as the
mean ± SD. Statistical comparisons were evaluated by one-way ANOVA.
Statistical significance was accepted at P<0.05.
Results
Determination of multidrug
resistance
Based on the CCK-8 method, it was observed that the
IC50 values of doxorubicin in the K562/ADM cells were
markedly increased in comparison with the values in the
drug-sensitive K562 cells. As indicated in Table I, the K562/ADM cells exhibited an
~38.55-fold increase in resistance to doxorubicin compared with the
parental K562 cells.
 | Table IDetermination of multidrug resistance
according to the sensitivity of K562/ADM and K562 cells toward
doxorubicin. |
Table I
Determination of multidrug resistance
according to the sensitivity of K562/ADM and K562 cells toward
doxorubicin.
| Treatment | K562/ADM
IC50 (µM) | K562
IC50 (µM) | Resistant fold |
|---|
| Doxorubicin | 90.60±1.28a | 2.35±0.49 | 38.55 |
Intrinsic cytotoxic effect of osthole in
the K562/ADM cells
To acquire an indication of the potential
therapeutic concentration as modulator, the CCK-8 assay was carried
out to evaluate the effect of osthole on K562/ADM cells. Osthole
inhibited the viability of the K562/ADM cells in a dose-dependent
manner in vitro (Fig. 1A).
As determined by the dose response curve, 5 µM osthole
exhibited no significant cytotoxicity (inhibition rate <5%)
while 15 µM osthole was weakly cytotoxic (inhibition rate
10–15%). Therefore, treatment concentrations of 5 and 15 µM
osthole were chosen in the following reversal experiments.
Reversal effect of osthole on the
sensitivity to doxorubicin in the K562/ADM cells
The influence of osthole on the sensitivity of
doxorubicin in the K562/ADM cells is shown in Table II and Fig. 1B. The reversal ability of the
modulator, valuated by RF, was scored as follows: RF>1 implies
enhanced drug sensitivity in the presence of osthole, RF=1 suggests
no effect and RF<1 indicates decreased drug sensitivity; the
greater the RF magnitude, the more powerful is the reversal effect
(30). Osthole enhanced the
sensitivity of the K562/ADM cells to doxorubicin by 1.79- and
3.84-fold at concentrations of 5 and 15 µM, respectively.
All these findings confirmed that osthole could enhance the potency
of doxorubicin against K562/ADM cells, supporting the notion that
osthole has a reversal effect on the drug sensitivity of K562/ADM
cells.
| Table IIEffect of osthole on the sensitivity
of the K562/ADM cells toward doxorubicin by CCK-8 assay. |
Table II
Effect of osthole on the sensitivity
of the K562/ADM cells toward doxorubicin by CCK-8 assay.
| Treatment | IC50
(µM) | RF |
|---|
| Doxorubicin
alone | 97.79±2.97 | |
| Doxorubicin + 5
µM osthole | 54.63±1.31a | 1.79 |
| Doxorubicin +15
µM osthole | 25.45±0.47a | 3.84 |
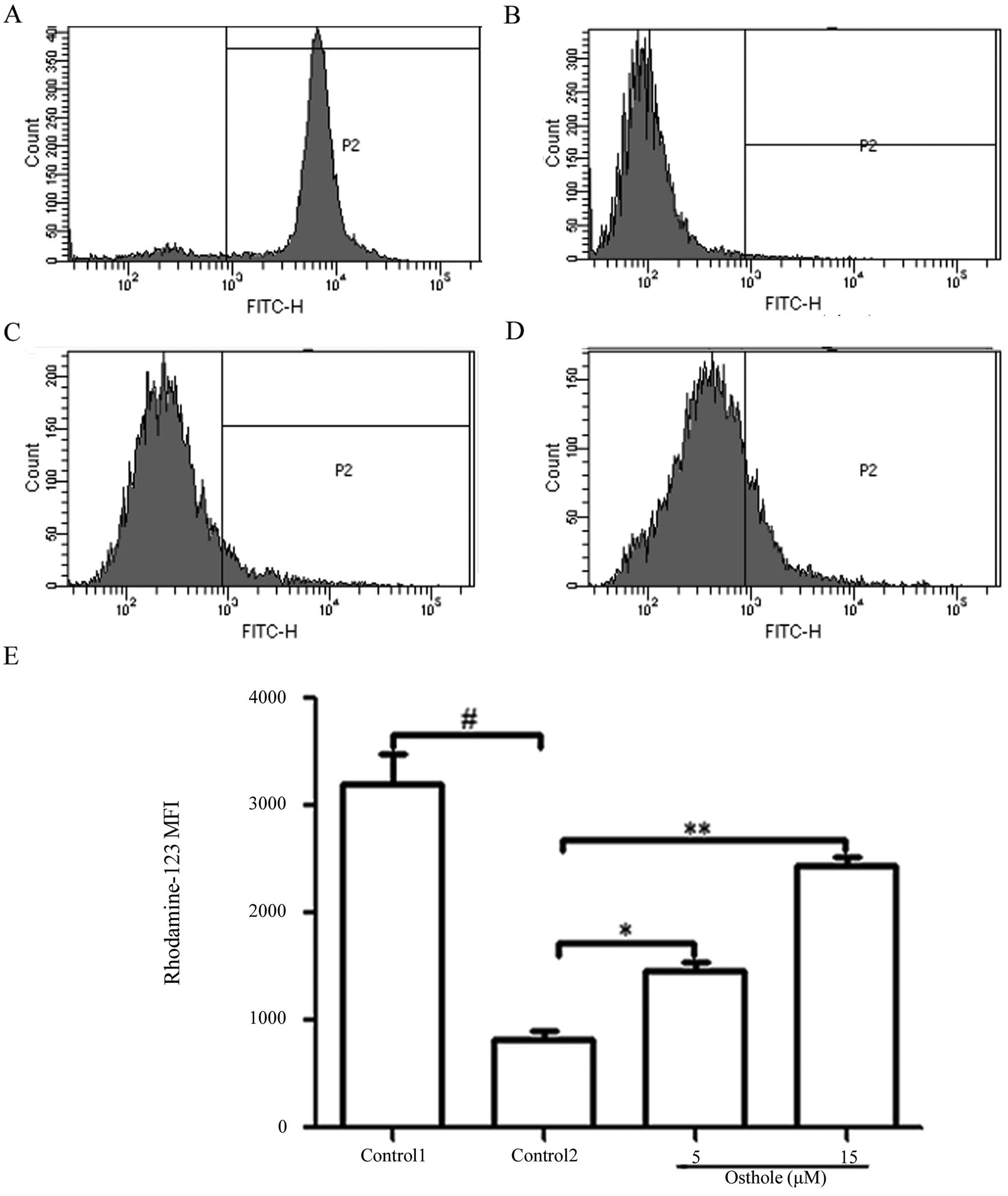
Enhancement of Rh-123 accumulation
Flow cytometry results revealed that the
intracellular accumulation of Rh-123 was evidently decreased in the
K562/ADM cells in contrast with the K562 cells and the MFI was
823±47.98 and 3209±101.95 (Fig. 2A and
B). Osthole (5 and 15 µM) enhanced the accumulation of
Rh-123 by 1.78- and 2.96-fold compared to the control group cells.
The MFI of Rh-123 was 1462±42.82 (osthole 5 µM) and
2437±46.47 (osthole 15 µM) (Fig.
2C–E).
Osthole increases the intracellular
accumulation of doxorubicin
The fluorescence characteristics of doxorubicin were
utilized to determine the drug concentration in the cells by flow
cytometry; intracellular MFI is representative of doxorubicin
concentration. The detection results signified that the MFI of K562
and K562/ADM cells was 1313±40.77 and 279±15.53 (Fig. 3A and B). The intracellular
accumulation of doxorubicin increased by 2.11- and 3.49-fold in
comparison with the negative control group in the K562/ADM cells
(Fig. 3C and D). These results
implied that osthole elevated the sensitivity of the K562/ADM cells
toward doxorubicin by increasing intracellular doxorubicin
accumulation.
Osthole efficiently decreases the P-gp
expression in the K562/ADM cells
K562/ADM cells express a higher level of P-gp than
K562 cells (31). As shown in
Fig. 4, after K562/ADM cells were
treated with different concentrations of osthole, the expression of
MDR1 at both the mRNA and protein levels was markedly decreased in
a dose-dependent manner.
Osthole inhibits PI3K/Akt signaling
pathway activity in the K562/ADM cells
According to our research, K562/ADM cells with
overexpressed P-gp demonstrated higher expression of the p-Akt
protein level than the parental K562 cells (Fig. 5A and B), which revealed that osthole
would be more effective in a resistant cell line when inhibiting
the PI3K/Akt signaling pathway. After treatment with osthole for 24
h, there was no apparent change in the total amount of Akt, whereas
p-Akt was reduced obviously (Fig. 5C
and D), which was parallel to the decrease in P-gp.
Osthole downregulates P-gp expression via
inhibition of the PI3K/Akt signaling pathway
To confirm the possibility that suppression of the
PI3K/Akt pathway results in decreased P-gp level, we investigated
the effect of the well-characterized pharmacological PI3K inhibitor
LY294002 on P-gp expression in the K562/ADM cells (32). Stimulation of K562/ADM cells with
LY294002 alone for 1 h led to inhibition of P-gp expression and Akt
phosphorylation. In addition, combined treatment with LY294002 and
osthole (pretreatment with 15 µM osthole for 24 h, then
treatment with LY294002 for 1 h) showed additive inhibitory effects
on P-gp expression and Akt phosphorylation (Fig. 6A and B). That is, the inhibition of
the PI3K/Akt pathway induced by osthole was responsible for the
reduced P-gp expression.
Further verification of the suppression
of P-gp expression via inhibition of the PI3K/Akt signaling pathway
by osthole
The PI3K-specific agonist insulin-like factor-1
(IGF-1) was used to further verify the relationship between
activation of the PI3K/Akt signaling pathway and P-gp expression
(33). As shown in Fig. 6C, the expression of p-Akt was
significantly increased by treatment with IGF-1 for 1 h. However,
the increasing trend was reversed by 15 µM osthole
(pretreatment with 15 µM osthole for 24 h, and then
treatment with IGF-1 for 1 h). The same trend was found in MDR1
expression at both the transcription and translation level. These
results further explain that osthole downregulated p-Akt activity,
resulting in the decreased expression of MDR1.
Discussion
Multidrug resistance (MDR) mediated by drug
transporters particularly P-gp has been a significant impediment to
the successful treatment of cancer (34). Hence, direct blockage of the P-gp
efflux pump or inhibition of its expression, thereby restoring the
sensitivity of tumor cells to chemotherapeutic agents, is an ideal
strategy for MDR reversal. To date, many P-gp inhibitors have been
developed. Verapamil and cyclosporine A, as classical
representatives of first-generation P-gp inhibitors, are combined
with various types of chemotherapy regimens for many neoplasms, but
the therapeutic effects are not satisfactory. Due to
pharmacokinetic interaction with chemotherapeutic drugs,
second-generation P-gp inhibitors such as dexverapamil, PSC 833 and
VX-710 are confined in clinical use (35). Time and resources have been spent on
the exploration of the third-generation P-gp inhibitors represented
by Zosuquidar (LY335979), Tariquidar (XR9576), Laniquidar
(R102933), Elacridar, PGP-4008, CBT-1 and HM30181. Despite the
initial enthusiasm following the preliminary report of XR9576,
clinical trials brought the tragic reality to light (36,37).
All in all, none of these hopeful modulators has been licensed for
application clinically owning to their toxicities at the high doses
needed for effective use or highly unacceptable side effects
(38). New reversal agents which
are efficient and minimally toxic require active exploration.
In order to find novel, non-toxic, and high affinity
P-gp inhibitors, more and more researchers have put emphasis on
screening active components from natural sources, which are
classified by some authors as fourth-generation P-gp inhibitors
(37). Osthole, a natural coumarin
compound, was verified to potently induce apoptosis and to
attenuate the migration of tumor cells but caused no significant
side effects to normal cells (23,24).
Furthermore, osthole is capable of enhancing the immunity of the
human body, which is a major advantage that makes it superior to
existing chemotherapy drugs (21).
Taken together, great expectations have been put on its potential
role in drug discovery and clinical utilization.
In the present study, we evaluated the efficacy of
osthole as a powerful reversal agent for overcoming drug resistance
of K562/ADM cells for the first time. Cytotoxicity analysis implied
that the cytotoxicity of osthole in K562/ADM cells was
dose-dependent. In accordance with the experimental results, weakly
cytotoxic concentrations of 5 and 15 µM osthole were
ultimately administered to the cells. Data indicated that
IC50 values of doxorubicin were significantly decreased
in the K562/ADM cells after osthole treatment, which indicated that
osthole could distinctly enhance the doxorubicin-induced
cytotoxicity toward K562/ADM cells. Herein, the reversal efficiency
of osthole was reflected by RF which reached ~4-fold. Moreover,
doxorubicin and rhodamine-123 efflux assay was exploited to assess
the inhibition of P-gp transport by osthole and to estimate the
interaction between osthole and this protein. The flow cytometric
assay indicated that osthole notably reversed the accumulation
deficit and blocked the cellular efflux of P-gp substrates
doxorubicin and rhoda-mine-123 in a concentration-dependent manner.
To reveal the mechanisms, the MDR1 expression at both the
transcription and translation levels were examined. The mRNA as
well as the protein expression levels of MDR1 were significantly
suppressed in the K562/ADM cells when treated with 5 and 15
µM osthole (Fig. 4). In
summery, osthole partially reversed MDR of K562/ADM cells by
inhibiting the cellular efflux function of P-gp and downregulating
the expression level of MDR1, thus elevating intracellular
chemotherapeutic accumulation.
Previous studies have indicated that the PI3K/Akt
pathway is excessively activated in a wide variety of hematologic
malignancies including CML, diffuse large B-cell lymphoma, acute
myeloid leukemia, T-cell acute lymphoblastic leukemia and chronic
lymphoblastic leukemia; it is considered to be responsible for
tumorigenesis by simultaneously promoting proliferation and
inhibiting apoptosis (39).
Morishita et al proposed that Akt phosphorylation can induce
chemotherapeutic resistance in B-pre-acute lymphoblastic leukemia
(40). Related studies suggest that
Akt is involved in the regulation of the MDR1 gene and that P-gp is
one of the key factors downstream of the PI3K/Akt pathway (41). To understand why osthole decreased
the expression level of MDR1, we further detected the
phosphorylation of Akt following treatment of osthole.
Interestingly, we found a parallel relationship between the
activity of Akt and expression of P-gp. In other words, the
expression level of p-Akt and P-gp were both decreased by addition
of osthole, but without a change in the levels of total Akt kinase.
Our findings are consistent with previous investigations in other
MDR tumor cells such as breast cancer cells, and gastric carcinoma
cells (19,42), which illustrate that excessive
activation of PI3K/Akt has an intimate correlation with P-gp
expression. Importantly, specific PI3K/Akt signaling pathway
inhibitor LY294002 and stimulator IGF-1 were used. In the LY294002
stimulating experiment, the inhibitory effect of osthole on p-Akt
and P-gp demonstrated in the LY294002 addition group was enhanced
in contrast with the combination of LY294002 and osthole group.
Moreover, IGF-1 was capable of escalating MDR1 expression and
promoting phosphorylation of Akt, while the trend was mostly
reversed by osthole. Thus, osthole reversed P-gp-mediated MDR by
inhibiting the PI3K/Akt signaling cascade. Unfortornately, it
should be noted that various alterations in the PI3K/Akt pathway
provide multiple molecular targets for treament and compounds the
difficulty to identify the key hub or hubs where osthole or other
targeted therapeutic interventions would be most efficacious.
Overall, we first revealed that osthole is a novel
and potent modulator which opens the way for reversing MDR in human
myelogenous leukemia K562/ADM cells. At the same time, the
potential association of the reversal of P-gp-mediated MDR and the
PI3K/Akt signaling cascade was identified, which may be the major
mechanism involved in the reversal of MDR by osthole in K562/ADM
cells.
Abbreviations:
|
P-gp
|
P-glycoprotein
|
|
PBS
|
phosphate-buffered saline
|
|
SDS-PAGE
|
sodium dodecyl sulphate-polyacrylamide
gel electrophoresis
|
|
MDR
|
multidrug resistance
|
|
MDR1
|
multidrug resistance gene 1
|
|
CML
|
chronic myeloid leukemia
|
Acknowledgments
This study was supported by the Shandong Science and
Technology Committee (no. 2010GSF10264), the Foundation of Shandong
Educational Committee (nos. J10LC60 and J11LC01), Natural Science
Foundation of Shandong Province (no. ZR2014HL032) and Projects of
Medical and Health Technology Development Program in Shandong
Province (no. 2014WS0183).
References
|
1
|
Mathisen MS, Kantarjian HM, Cortes J and
Jabbour E: Mutant BCR-ABL clones in chronic myeloid leukemia.
Haematologica. 96:347–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cao R, Wang Y and Huang N: Discovery of
2-acylami-nothiophene-3-carboxamides as multitarget inhibitors for
BCR-ABL kinase and microtubules. J Chem Inf Model. 55:2435–2442.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
de Lavallade H, Apperley JF, Khorashad JS,
Milojkovic D, Reid AG, Bua M, Szydlo R, Olavarria E, Kaeda J,
Goldman JM, et al: Imatinib for newly diagnosed patients with
chronic myeloid leukemia: Incidence of sustained responses in an
intention-to-treat analysis. J Clin Oncol. 26:3358–3363. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mahon FX, Hayette S, Lagarde V, Belloc F,
Turcq B, Nicolini F, Belanger C, Manley PW, Leroy C, Etienne G, et
al: Evidence that resistance to nilotinib may be due to BCR-ABL,
Pgp, or Src kinase overexpression. Cancer Res. 68:9809–9816. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wei YL, Xu L, Liang Y, Xu XH and Zhao XY:
Berbamine exhibits potent antitumor effects on imatinib-resistant
CML cells in vitro and in vivo. Acta Pharmacol Sin. 30:451–457.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Corrêa S, Binato R, Du Rocher B,
Castelo-Branco MT, Pizzatti L and Abdelhay E: Wnt/β-catenin pathway
regulates ABCB1 transcription in chronic myeloid leukemia. BMC
Cancer. 12:3032012. View Article : Google Scholar
|
|
7
|
Souza PS, Vasconcelos FC, De Souza Reis
FR, Nestal De Moraes G and Maia RC: P-glycoprotein and survivin
simultaneously regulate vincristine-induced apoptosis in chronic
myeloid leukemia cells. Int J Oncol. 39:925–933. 2011.PubMed/NCBI
|
|
8
|
Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE
and Gottesman MM: P-glycoprotein: From genomics to mechanism.
Oncogene. 22:7468–7485. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dulucq S, Bouchet S, Turcq B, Lippert E,
Etienne G, Reiffers J, Molimard M, Krajinovic M and Mahon FX:
Multidrug resistance gene (MDR1) polymorphisms are associated with
major molecular responses to standard-dose imatinib in chronic
myeloid leukemia. Blood. 112:2024–2027. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cao H, Dronadula N and Rao GN: Thrombin
induces expression of FGF-2 via activation of PI3K-Akt-Fra-1
signaling axis leading to DNA synthesis and motility in vascular
smooth muscle cells. Am J Physiol Cell Physiol. 290:C172–C182.
2006. View Article : Google Scholar
|
|
12
|
Hales EC, Taub JW and Matherly LH: New
insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling
axis: Targeted therapy of γ-secretase inhibitor resistant T-cell
acute lymphoblastic leukemia. Cell Signal. 26:149–161. 2014.
View Article : Google Scholar
|
|
13
|
Kumar N, Afeyan R, Sheppard S, Harms B and
Lauffenburger DA: Quantitative analysis of Akt phosphorylation and
activity in response to EGF and insulin treatment. Biochem Biophys
Res Commun. 354:14–20. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang H, Bajraszewski N, Wu E, Wang H,
Moseman AP, Dabora SL, Griffin JD and Kwiatkowski DJ: PDGFRs are
critical for PI3K/Akt activation and negatively regulated by mTOR.
J Clin Invest. 117:730–738. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
García MG, Alaniz LD, Cordo Russo RI,
Alvarez E and Hajos SE: PI3K/Akt inhibition modulates multidrug
resistance and activates NF-kappaB in murine lymphoma cell lines.
Leuk Res. 33:288–296. 2009. View Article : Google Scholar
|
|
16
|
Chiarini F, Del Sole M, Mongiorgi S,
Gaboardi GC, Cappellini A, Mantovani I, Follo MY, McCubrey JA and
Martelli AM: The novel Akt inhibitor, perifosine, induces
caspase-dependent apoptosis and downregulates P-glycoprotein
expression in multidrug-resistant human T-acute leukemia cells by a
JNK-dependent mechanism. Leukemia. 22:1106–1116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xie X, Tang B, Zhou J, Gao Q and Zhang P:
Inhibition of the PI3K/Akt pathway increases the chemosensitivity
of gastric cancer to vincristine. Oncol Rep. 30:773–782.
2013.PubMed/NCBI
|
|
18
|
Cheng L, Luo S, Jin C, Ma H, Zhou H and
Jia L: FUT family mediates the multidrug resistance of human
hepatocellular carcinoma via the PI3K/Akt signaling pathway. Cell
Death Dis. 4:e9232013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mao Z, Zhou J, Luan J, Sheng W, Shen X and
Dong X: Tamoxifen reduces P-gp-mediated multidrug resistance via
inhibiting the PI3K/Akt signaling pathway in ER-negative human
gastric cancer cells. Biomed Pharmacother. 68:179–183. 2014.
View Article : Google Scholar
|
|
20
|
Tazzari PL, Cappellini A, Ricci F,
Evangelisti C, Papa V, Grafone T, Martinelli G, Conte R, Cocco L,
McCubrey JA, et al: Multidrug resistance-associated protein 1
expression is under the control of the phosphoinositide 3
kinase/Akt signal transduction network in human acute myelogenous
leukemia blasts. Leukemia. 21:427–438. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang HY, Hsu YF, Chiu PT, Ho SJ, Wang CH,
Chi CC, Huang YH, Lee CF, Li YS, Ou G, et al: Anti-cancer activity
of an osthole derivative, NBM-T-BMX-OS01: Targeting vascular
endothelial growth factor receptor signaling and angiogenesis. PLoS
One. 8:e815922013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu Y, Wen Q, Liang W, Kang T, Ren L, Zhang
N, Zhao D, Sun D and Yang J: Osthole reverses beta-amyloid peptide
cytotoxicity on neural cells by enhancing cyclic AMP response
element-binding protein phosphorylation. Biol Pharm Bull.
36:1950–1958. 2013. View Article : Google Scholar
|
|
23
|
Chou SY, Hsu CS, Wang KT, Wang MC and Wang
CC: Antitumor effects of osthol from Cnidium monnieri: An in vitro
and in vivo study. Phytother Res. 21:226–230. 2007. View Article : Google Scholar
|
|
24
|
Jarząb A, Grabarska A, Kiełbus M,
Jeleniewicz W, Dmoszyńska-Graniczka M, Skalicka-Woźniak K,
Sieniawska E, Polberg K and Stepulak A: Osthole induces apoptosis,
suppresses cell-cycle progression and proliferation of cancer
cells. Anticancer Res. 34:6473–6480. 2014.
|
|
25
|
Liu LY, Huang WJ, Ho FM, Lin RJ, Lin SY,
Suk FM and Liang YC: N-Hydroxycinnamide derivatives of osthole
inhibit cell migration and invasion by suppressing Smad2 and Akt
pathways in human colorectal adenocarcinoma cells. Chem Biol
Interact. 217:1–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tsai CF, Yeh WL, Chen JH, Lin C, Huang SS
and Lu DY: Osthole suppresses the migratory ability of human
glioblastoma multiforme cells via inhibition of focal adhesion
kinase-mediated matrix metalloproteinase-13 expression. Int J Mol
Sci. 15:3889–3903. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu XM, Zhang Y, Qu D, Feng XW, Chen Y and
Zhao L: Osthole suppresses migration and invasion of A549 human
lung cancer cells through inhibition of matrix metalloproteinase-2
and matrix metallopeptidase-9 in vitro. Mol Med Rep. 6:1018–1022.
2012.PubMed/NCBI
|
|
28
|
Ye Y, Han X, Guo B, Sun Z and Liu S:
Combination treatment with platycodin D and osthole inhibits cell
proliferation and invasion in mammary carcinoma cell lines. Environ
Toxicol Pharmacol. 36:115–124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang L, Jiang G, Yao F, He Y, Liang G,
Zhang Y, Hu B, Wu Y, Li Y and Liu H: Growth inhibition and
apoptosis induced by osthole, a natural coumarin, in hepatocellular
carcinoma. PLoS One. 7:e378652012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang HY, Zhao L, Yang Z, Zhao Q, Qiang L,
Ha J, Li ZY, You QD and Guo QL: Oroxylin A reverses multi-drug
resistance of human hepatoma BEL7402/5-FU cells via downregulation
of P-glycoprotein expression by inhibiting NF-κB signaling pathway.
Mol Carcinog. 51:185–195. 2012. View
Article : Google Scholar
|
|
31
|
Xu X, Zhang Y, Li W, Miao H, Zhang H, Zhou
Y, Li Z, You Q, Zhao L and Guo Q: Wogonin reverses multi-drug
resistance of human myelogenous leukemia K562/A02 cells via
downregulation of MRP1 expression by inhibiting Nrf2/ARE signaling
pathway. Biochem Pharmacol. 92:220–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barancík M, Bohácová V, Sedlák J, Sulová Z
and Breier A: LY294,002, a specific inhibitor of PI3K/Akt kinase
pathway, antagonizes P-glycoprotein-mediated multidrug resistance.
Eur J Pharm Sci. 29:426–434. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lau MT and Leung PC: The PI3K/Akt/mTOR
signaling pathway mediates insulin-like growth factor 1-induced
E-cadherin down-regulation and cell proliferation in ovarian cancer
cells. Cancer Lett. 326:191–198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li X, Sun B, Zhu CJ, Yuan HQ, Shi YQ, Gao
J, Li SJ and Lou HX: Reversal of p-glycoprotein-mediated multidrug
resistance by macrocyclic bisbibenzyl derivatives in
adriamycin-resistant human myelogenous leukemia (K562/A02) cells.
Toxicol In Vitro. 23:29–36. 2009. View Article : Google Scholar
|
|
35
|
Zhu H, Liu Z, Tang L, Liu J, Zhou M, Xie
F, Wang Z, Wang Y, Shen S, Hu L, et al: Reversal of P-gp and
MRP1-mediated multidrug resistance by H6, a gypenoside aglycon from
Gynostemma pentaphyllum, in vincristine-resistant human oral cancer
(KB/VCR) cells. Eur J Pharmacol. 696:43–53. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim TE, Lee H, Lim KS, Lee S, Yoon SH,
Park KM, Han H, Shin SG, Jang IJ, Yu KS, et al: Effects of HM30181,
a P-glycoprotein inhibitor, on the pharmacokinetics and
pharmacodynamics of loperamide in healthy volunteers. Br J Clin
Pharmacol. 78:556–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Palmeira A, Sousa E, Vasconcelos MH and
Pinto MM: Three decades of P-gp inhibitors: Skimming through
several generations and scaffolds. Curr Med Chem. 19:1946–2025.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sprachman MM, Laughney AM, Kohler RH and
Weissleder R: In vivo imaging of multidrug resistance using a third
generation MDR1 inhibitor. Bioconjug Chem. 25:1137–1142. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang X, Dong W, Zhou H, Li H, Wang N,
Miao X and Jia L: α-2,8-Sialyltransferase is involved in the
development of multidrug resistance via PI3K/Akt pathway in human
chronic myeloid leukemia. IUBMB Life. 67:77–87. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Morishita N, Tsukahara H, Chayama K,
Ishida T, Washio K, Miyamura T, Yamashita N, Oda M and Morishima T:
Activation of Akt is associated with poor prognosis and
chemotherapeutic resistance in pediatric B-precursor acute
lymphoblastic leukemia. Pediatr Blood Cancer. 59:83–89. 2012.
View Article : Google Scholar
|
|
41
|
Han Z, Hong L, Han Y, Wu K, Han S, Shen H,
Li C, Yao L, Qiao T and Fan D: Phospho Akt mediates multidrug
resistance of gastric cancer cells through regulation of P-gp,
Bcl-2 and Bax. J Exp Clin Cancer Res. 26:261–268. 2007.PubMed/NCBI
|
|
42
|
Lin X, Zhang X, Wang Q, Li J, Zhang P,
Zhao M and Li X: Perifosine downregulates MDR1 gene expression and
reverses multidrug-resistant phenotype by inhibiting PI3K/Akt/NF-κB
signaling pathway in a human breast cancer cell line. Neoplasma.
59:248–256. 2012. View Article : Google Scholar
|