Introduction
Pancreatic cancer is one of the most aggressive
malignancies and is associated with high mortality and a 5-year
survival rate of ~5% (1). The
lethality of pancreatic cancer is characterized by rapid invasion
of the surrounding tissue, early metastatic disease, and poor
responses to standard chemotherapy and radiotherapy (2). Surgical resection and chemotherapy
regimens, which include gemcitabine, currently provide the best
clinical benefits. Gemcitabine has been the reference drug for the
treatment of this often fatal disease for more than 10 years
(3). A number of new anticancer
drugs have been introduced in the last decade with the aim of
improving the survival of pancreatic cancer patients. Despite
continuous efforts to develop new agents, none of the currently
available chemotherapeutic agents have an objective response rate
higher than 10% (4,5). Therefore, there is an urgent need to
develop better therapeutic strategies for pancreatic cancer.
Several recent studies suggest that dietary
phytochemicals offer chemopreventive effects against many types of
malignancies (6). Furthermore,
epidemiological evidence revealed an inverse relationship between
the intake amount of dietary isothiocyanates (ITCs) and cancer risk
(7,8). Among all ITCs, phenethyl
isothiocyanate (PEITC) has reached the level of phase 2 clinical
trials for lung and oral cancer prevention (9,10).
PEITC is a well-known phytochemical found in its glucosinolate
precursor form in cruciferous vegetables such as watercress and
broccoli (9,11). Due to its potential use in
preventive medicine and as a cancer therapeutic agent, PEITC has
attained great significance as an anticancer agent (12). PEITC has been shown to inhibit
cancer cell growth, survival, and angiogenesis in many cancer cell
lines (9). Furthermore, it is a
potent generator of reactive oxygen species (ROS) (13,14).
Drug screening studies recently discovered that Ras transformation
renders cells sensitive to ROS-induced, non-apoptotic,
iron-dependent mode of cell death (15,16).
This mode of programmed necrosis, termed ferroptosis, is
characterized by the loss of redox homeostasis, increased lipid
peroxidation, and inhibition by the small molecule ferrostatin-1
(Ferr-1) or iron chelator deferoxamine (17,18).
However, PEITC-induced ROS-ferroptosis pathway-mediated pancreatic
cancer cell death has not yet been evaluated.
We previously determined whether inducers of
differentiation in leukemia cells also have the ability to control
the growth of solid tumors. Cotylenin A (CN-A), which is a
fucicoccan-diterpene glycoside with a complex sugar moiety, was
originally isolated as a plant growth regulator and has been shown
to affect several physiological processes in higher plants
(19). We previously reported that
CN-A exhibits potent differentiation-inducing activity in several
human and murine myeloid leukemia cell lines and in leukemia cells
freshly isolated from patients with acute myeloid leukemia
(20–23). We recently demonstrated that CN-A
significantly potentiated the arsenic trioxide-induced inhibition
of cell growth in a liquid culture, and arsenic trioxide-induced
inhibition of anchorage-independent growth in a semisolid culture
in human breast cancer cells (24).
Furthermore, we found that the pretreatment with
N-acetylcysteine, a typical ROS scavenger, significantly
reduced combination treatment-induced cell growth inhibition
(24). These findings suggest that
oxidative responses are important events in the corporative
inhibition of the growth of cancer cells induced by the combined
treatment with CN-A and arsenic trioxide. We then hypothesized that
CN-A-induced antitumor activity against solid tumors including
breast and pancreatic cancer is enhanced by ROS inducers. In the
present study, we found that CN-A and PEITC, as a model ROS inducer
to test this hypothesis, synergistically inhibited the
proliferation of MIAPaCa-2, PANC-1 and gemcitabine-resistant PANC-1
cells. Our results also suggest that synergistic cell death induced
by the combined treatment with CN-A and PEITC is mainly due to the
induction of ferroptosis, while CN-A or PEITC alone cannot induce
ferroptosis in pancreatic cancer cells.
Materials and methods
Cell culture
The human pancreatic cancer cell lines MIAPaCa-2,
PANC-1 and CFPAC-1 were purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA). PANC-1/GR cells were obtained
after a long-term culture in the presence of gemcitabine. These
pancreatic cancer cells were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS) at 37°C in a
humidified atmosphere of 5% carbon dioxide in air.
Materials
CN-A, ISIR-042, ISIR-005 and fusicoccin J (FC-J)
were prepared as previously described (19,25).
PEITC, gemcitabine,
3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetra-zolium bromide (MTT),
N-acetyl-L-cysteine (NAC), trolox, Ferr-1, 3-methyladenine
(3-MA), deferoxamine mesylate (DFO), Mito-TEMPO, RPMI-1640 medium
and DMEM/F12 medium were purchased from Sigma-Aldrich (St. Louis,
MO, USA). 5-(and-6)-Chloromethyl-2′,7′-dichlorodihydro-fluorescein
diacetate (CM-H2DCFDA), an acetyl ester, was obtained
from Invitrogen (Carlsbad, CA, USA). Z-VAD-FMK and liproxstatin
were purchased from Selleckchem (Houston, TX, USA). Q-VD-OPH (QVD)
was obtained from Tonbo Biosciences (San Diego, CA, USA).
Chloroquine (CQ) and methyl cellulose were purchased from Wako
(Osaka, Japan). Necrostatin-1s (Nec-1s) was obtained from BioVision
(Milpitas, CA, USA).
Cell growth and viability
Cells were seeded at 1–3×104 cells/ml
into a 24-well multidish. After being cultured with or without test
compounds for the indicated times, viable cells were examined by a
modified MTT assay (26).
Assay of anchorage-independent
growth
MIAPaCa-2 or PANC-1 cells (2×103
cells/well) were plated in RPMI-1640 medium supplemented with 10%
FBS and 1.0% methylcellulose in a 24-well ultra-low attachment
multidish (Corning, Corning, NY, USA). Colonies containing 10 or
more cells were counted 12 days after seeding.
Measurement of ROS generation
The production of ROS was monitored by flow
cytometry with CM-H2DCFDA (a derivative of DCF-DA) as a
probe. Briefly, MIAPaCa-2 cells treated with CN-A and/or PEITC were
incubated with 10 μM CM-H2DCFDA in
phosphate-buffered saline (PBS) for 30 min. After removing the
probe, the cells were incubated in warm culture medium for 10 min.
The medium was removed, and the cells were detached with a brief
treatment of 0.05% trypsin-EDTA solution. After the addition of
fresh culture medium, the cells were collected by centrifugation,
washed once with PBS and suspended in PBS. Fluorescence was
monitored using a BD FACSCanto II flow cytometer (BD Biosciences,
San Jose, CA, USA).
Statistical analysis
Values were compared using a two-tailed Student's
t-test. Differences between the means were considered significant
when P-values were <0.05.
Results
CN-A and PEITC synergistically inhibit
the growth of human pancreatic cancer MIAPaCa-2 and PANC-1
cells
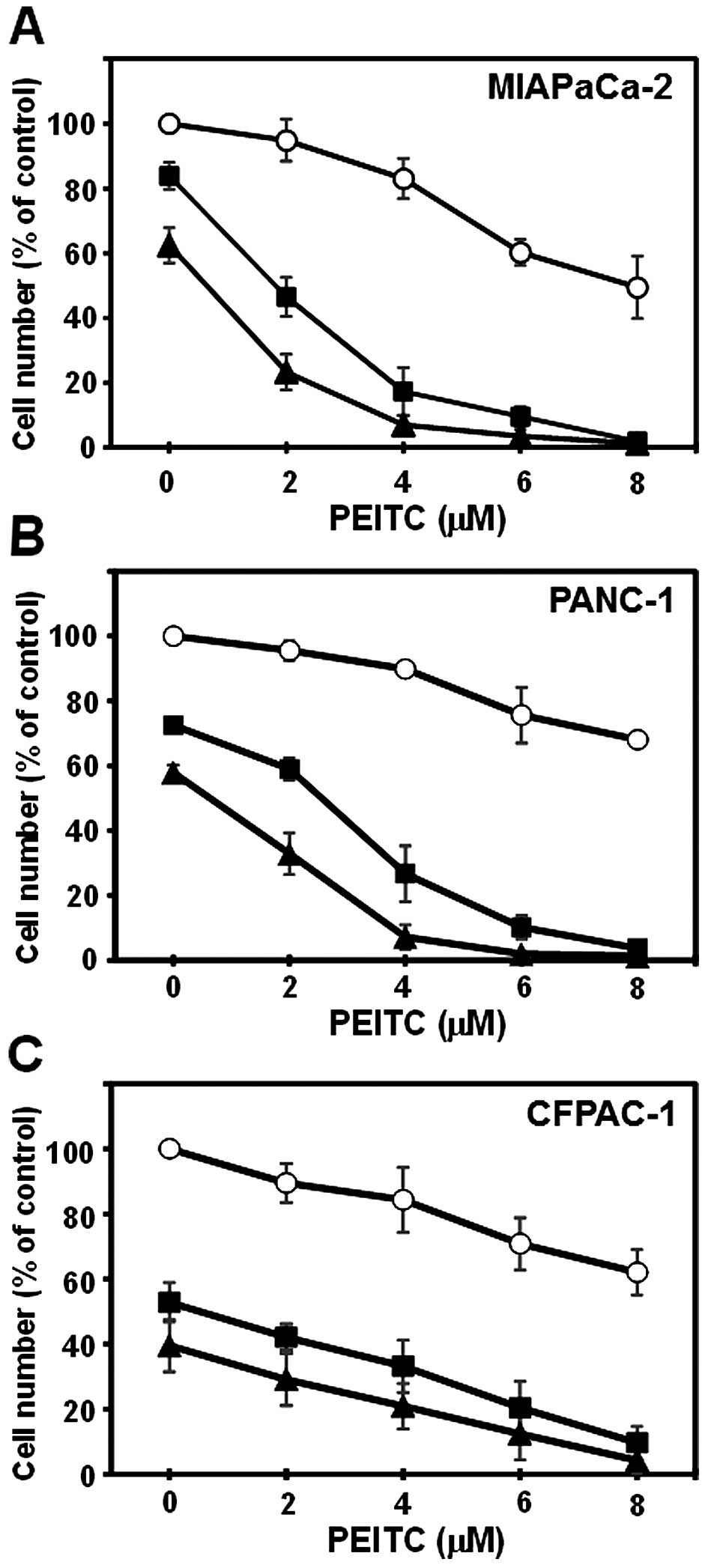
MIAPaCa-2, PANC-1 and CFPAC-1 cells were cultured
with or without CN-A, PEITC or CN-A plus PEITC for 5 days. CN-A and
PEITC synergistically inhibited the proliferation of MIAPaCa-2 and
PANC-1 cells (Fig. 1A and B).
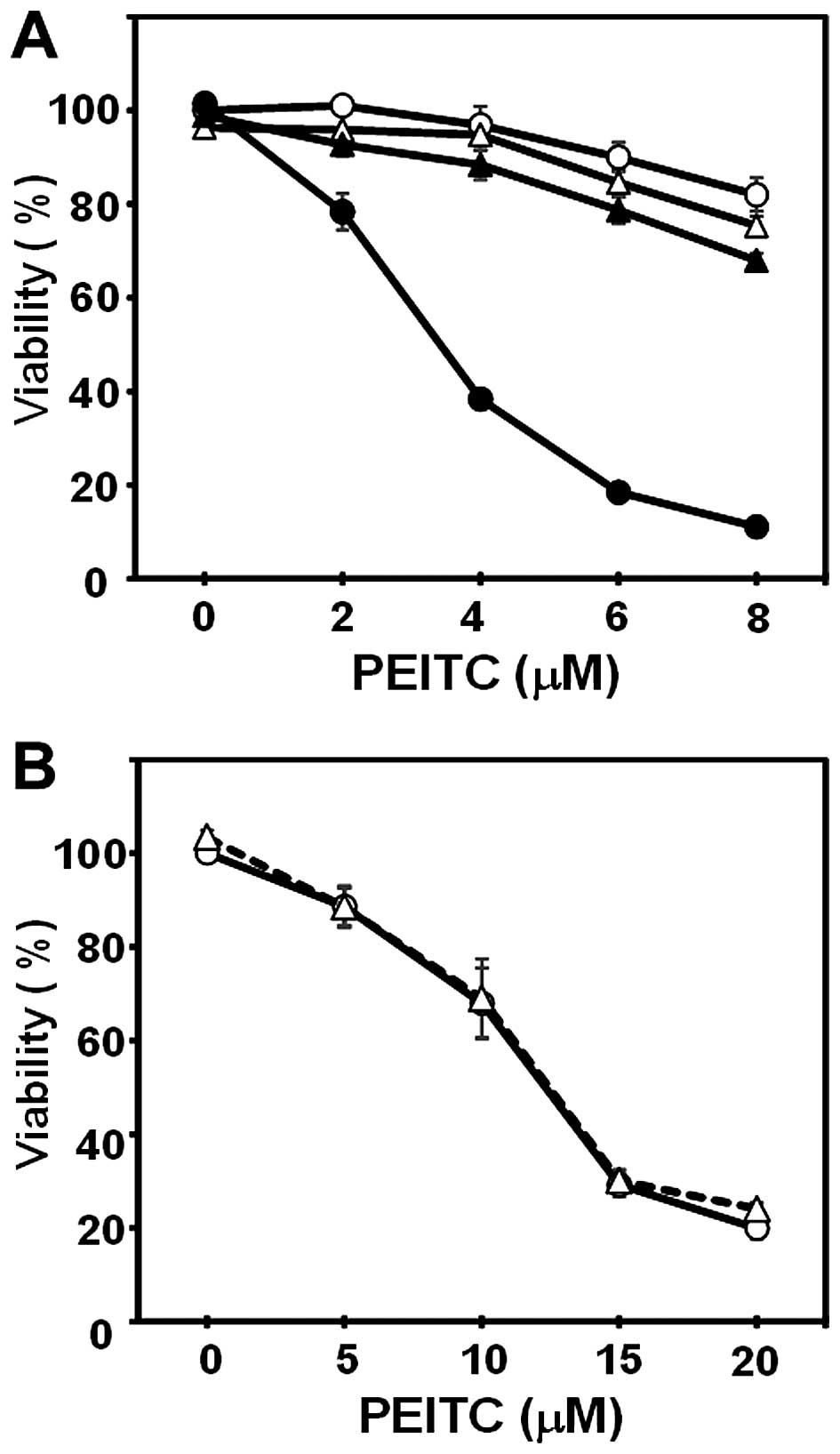
Although PEITC alone, even at a higher concentration (8 μM),
inhibited the growth of MIAPaCa-2 cells to ~50% of the control and
CN-A (10 μ/ml) alone slightly inhibited the growth of
MIAPaCa-2 cells, in the presence of CN-A (10 μ/ml), PEITC at
2, 4 or 6 μM inhibited the growth of MIAPaCa-2 cells to ~50,
~20 or ~10% of the control, respectively (Fig. 1A). Similar synergistic growth
inhibition induced by CN-A plus PEITC was observed in PANC-1 cells
(Fig. 1B). CN-A and PEITC
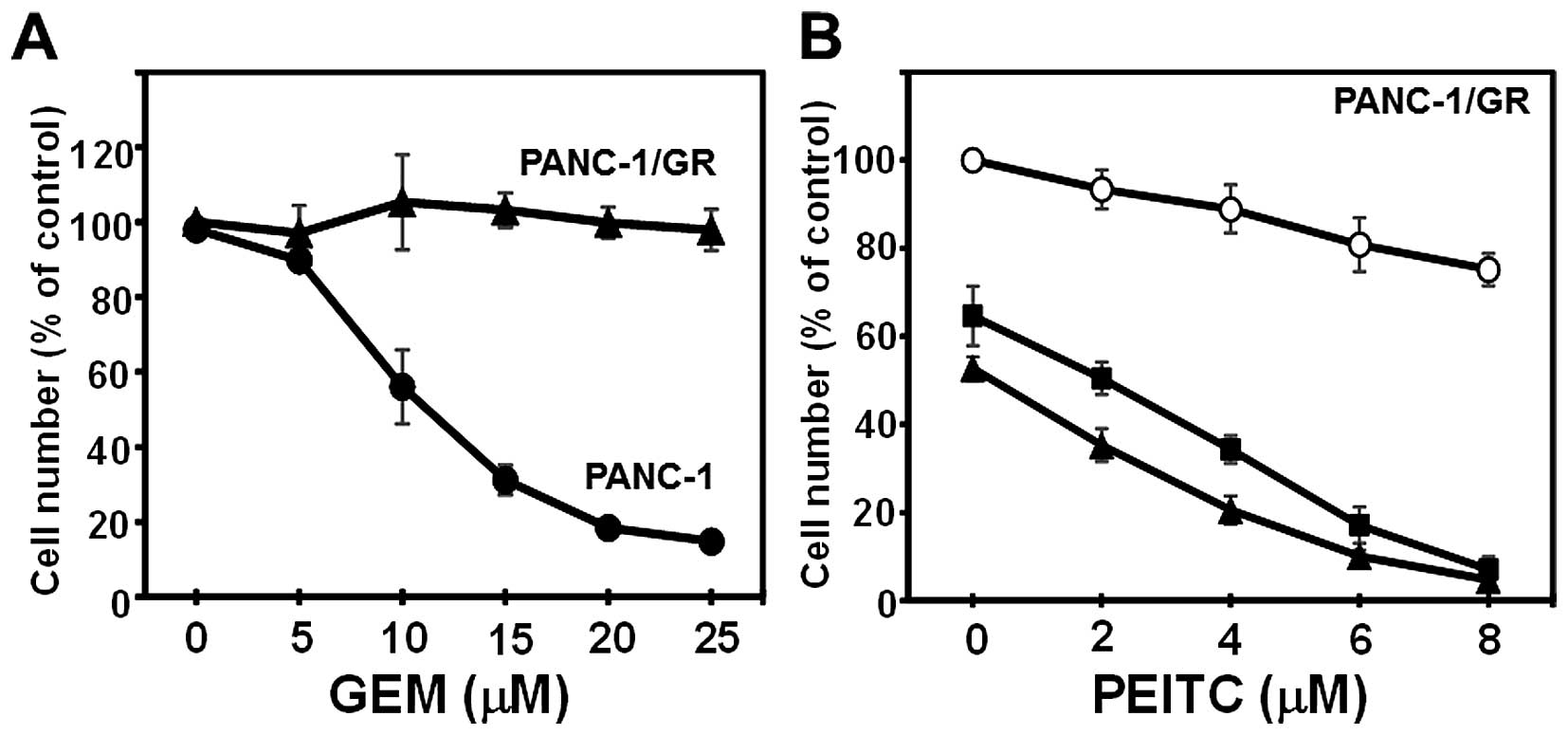
additively inhibited the proliferation of CFPAC-1 cells (Fig. 1C). In order to determine whether
CN-A and PEITC inhibit the proliferation of chemotherapeutic
drug-resistant pancreatic cancer cells, we obtained
gemcitabine-resistant PANC-1 cells (PANC-1/GR) after a long-term
culture in the presence of gemcitabine (final concentration at 100
μM). Although the proliferation of the parental PANC-1 cells
was dose-dependently inhibited by gemcitabine, the proliferation of
PANC-1/GR cells was not inhibited by gemcitabine, even at 25
μM (Fig. 2A). CN-A and PEITC
also synergistically inhibited the proliferation of the PANC-1/GR
cells (Fig. 2B).
CN-A and PEITC synergistically inhibit
the anchorage-independent growth of MIAPaCa-2 cells
Since anchorage-independent growth has been
correlated with tumorigenic potential, we next investigated whether
this combined treatment with CN-A and PEITC effectively inhibits
the anchorage-independent growth of these pancreatic cancer cells.
Although CN-A (10 μg/ml) or PEITC (5 μM) alone
inhibited colony formation by MIAPaCa-2 cells to ~60% of the
control, the combined treatment with CN-A and PEITC inhibited
colony formation to <5% of the control (Fig. 3A and B). CN-A and PEITC also
synergistically inhibited the anchorage-independent growth of the
PANC-1 cells (data not shown).
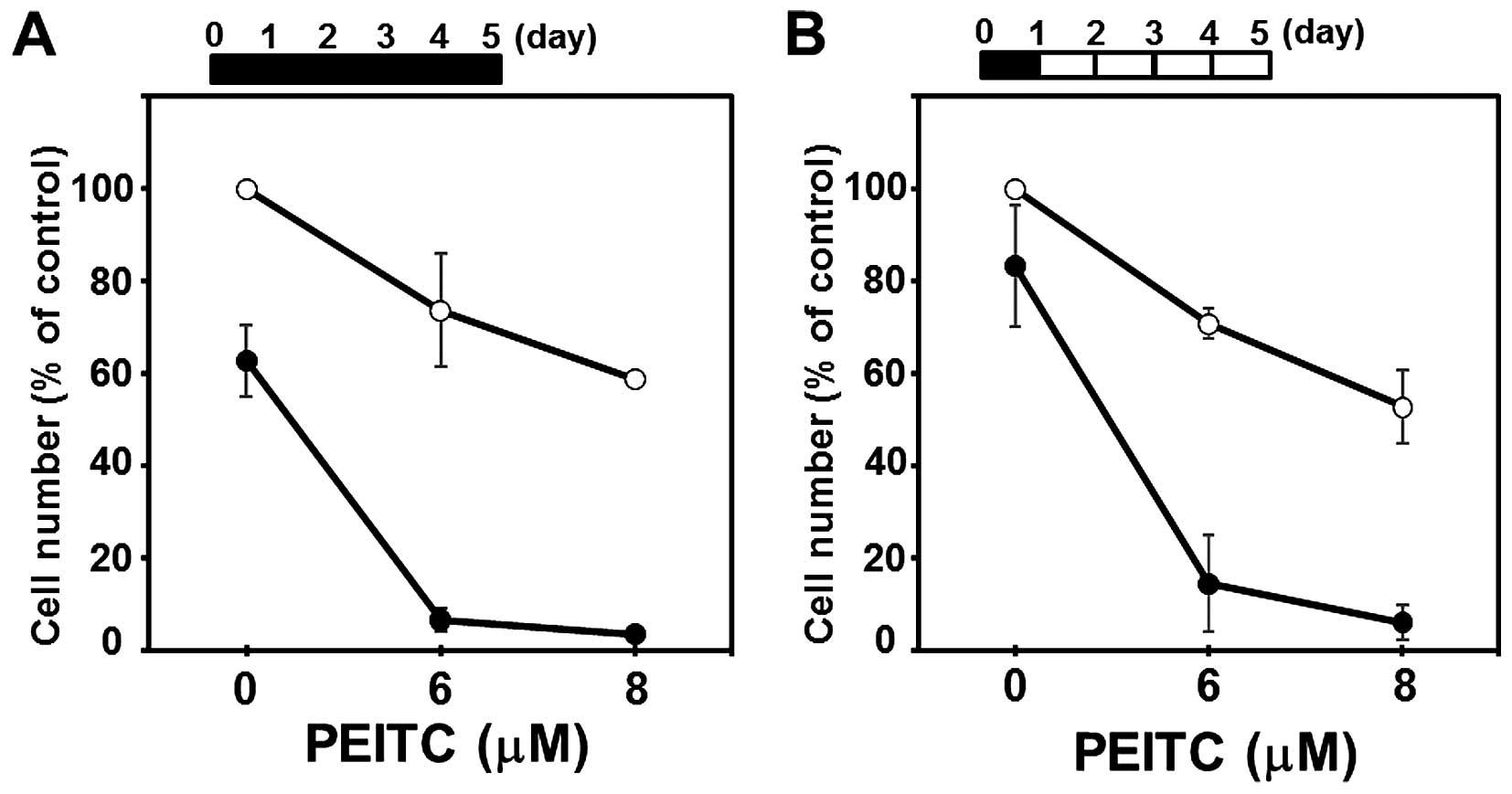
Combined treatment with CN-A and PEITC
strongly induces cell death in the MIAPaCa-2 and PANC-1 cells after
a short-term culture
Although a 1-day treatment with CN-A (5–20
μg/ml) or PEITC (2–8 μM) alone did not markedly
affect the viability of the MIAPaCa-2 and PANC-1 cells, the
combined treatment with CN-A and PEITC for 1 day strongly induced
cell death in the MIAPaCa-2 and PANC-1 cells (Figs. 4, 6A
and 8; data not shown). We
determined whether the effects of the combined treatment with CN-A
and PEITC for 1 day were sufficient to exert synergistic growth
inhibition in a 5-day culture as described above (Fig. 1A and B). After PANC-1 cells were
treated with CN-A (15 μg/ml) plus PEITC (6 or 8 μM)
for 1 day, PANC-1 cells were washed and further cultured for
another 4 days without CN-A or PEITC (Fig. 5B). At 5 days, the number of PANC-1
cells after the combined treatment with CN-A plus PEITC for only 1
day was almost similar to that induced by the continuous 5-day
treatment with CN-A plus PEITC (Fig.
5A).
Effects of CN-A analogues on the
viability of PANC-1 cells in the presence of PEITC
We next examined whether CN-A analogues and PEITC
also cooperatively reduce the viability of PANC-1 cells after a
short-term treatment. We previously reported that the synthetic
CN-A-derivatives ISIR-005 and ISIR-042, as well as CN-A in
combination with certain bioactive agents such as rapamycin and
arsenic trioxide synergistically induced antitumor activities in
several types of cancer cells, and that FC-J (a CN-A-related
natural product) did not exert such synergistic effects (24,25,27,28).
As shown in Fig. 6B–D, neither
ISIR-005, ISIR-042 nor FC-J reduced cell viability, even in the
presence of 8 μM PEITC for 16 h; however, the combined
treatment with CN-A and PEITC cooperatively reduced the viability
of the PANC-1 cells (Fig. 6A).
These results suggest that the mechanism underlying the synergistic
induction of cell death induced by CN-A plus PEITC differed from
the other synergistic combination effects with CN-A, ISIR-005, or
ISIR-042 plus other agents as previously reported (24,25,27,28).
CN-A and PEITC synergistically trigger
ROS accumulation in pancreatic cancer cells
Previous studies demonstrated that increased ROS
levels are responsible for the cell death induced by PEITC
(9,12–14).
We recently reported that the combined treatment with CN-A plus
arsenic trioxide-induced growth inhibition in breast cancer MCF-7
cells was significantly reduced by pretreatment with the
antioxidant compound NAC (24).
Therefore, we measured ROS levels in CN-A plus PEITC-treated
pancreatic cancer cells. We evaluated the effects of the combined
treatment with CN-A and PEITC on ROS generation in the MIAPaCa-2
and PANC-1 cells by flow cytometry using 10 μM
CM-H2DCFDA. MIAPaCa-2 cells were treated with 20
μg/ml CN-A and/or 4 μM PEITC for 4 h. As expected,
PEITC-treated MIAPaCa-2 cells exhibited a significant increase
(5-fold) in the basal ROS content. We found that the treatment with
CN-A plus PEITC resulted in a marked increase (~20-fold) in ROS
generation, whereas treatment with CN-A alone resulted in a
2.3-fold increase in ROS generation (Fig. 7). Similar results were obtained from
PANC-1 cells treated with CN-A plus PEITC (data not shown).
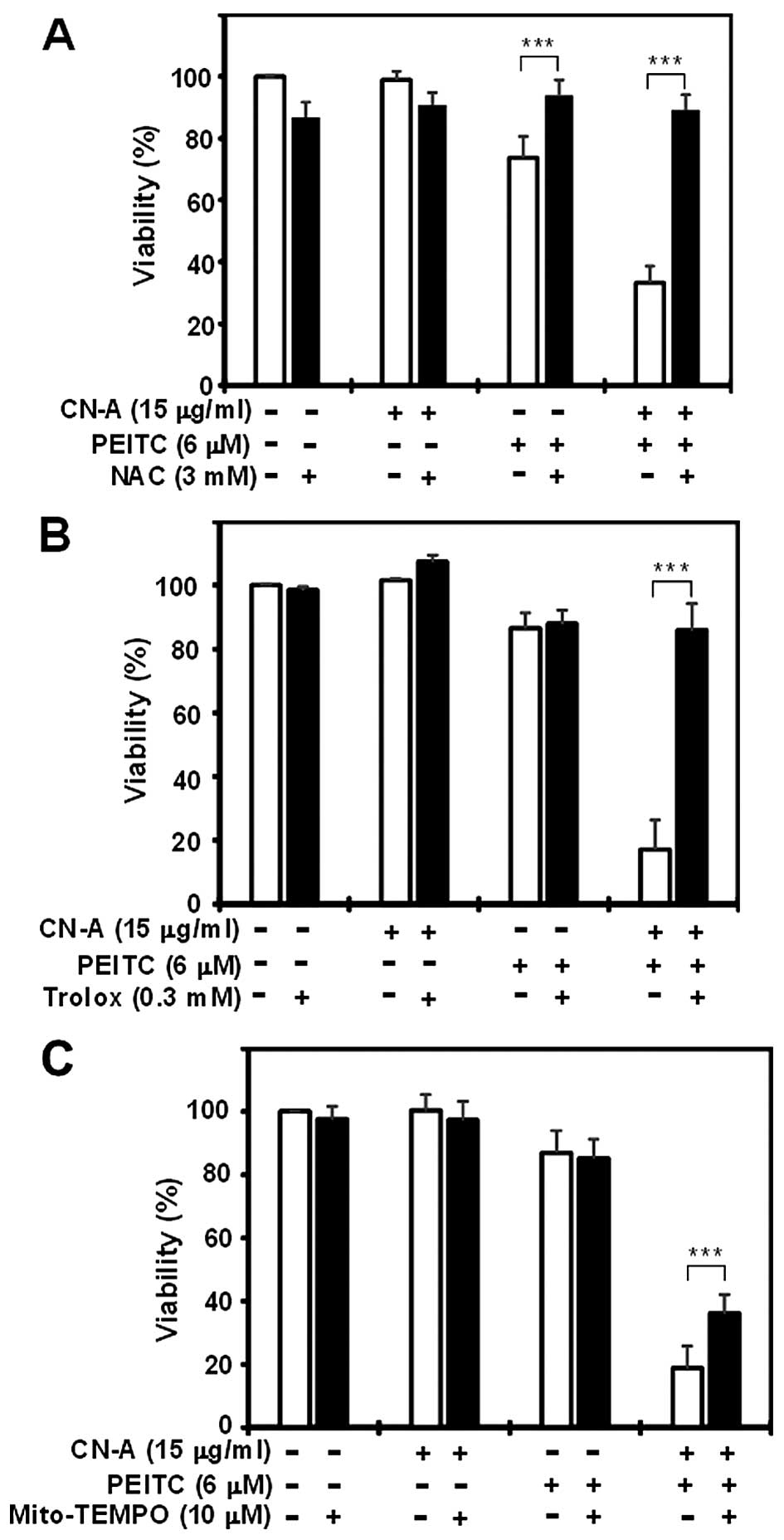
ROS scavengers, NAC and trolox reverse
CN-A plus PEITC-induced cell death in pancreatic cancer cells
We subsequently determined whether pancreatic cancer
cell death induced by the combined treatment with CN-A and PEITC
was dependent on ROS generation and mitochondrial ROS. At 16 h, the
CN-A (15 μg/ml) plus PEITC (6 μM) treatment markedly
decreased PANC-1 cell viability to ~20–30% of the control, while
CN-A alone did not significantly reduce cell viability and PEITC
alone reduced viability to ~80% of the control (Fig. 8). The ROS scavenger NAC (3 mM)
almost completely rescued the reduction in cell viability induced
by CN-A plus PEITC (Fig. 8A).
Similar results were obtained in MIAPaCa-2 cells (data not shown).
Furthermore, another ROS scavenger, trolox (0.3 mM) also canceled
the reduction in cell viability induced by CN-A plus PEITC
(Fig. 8B). In contrast, the
mitochondrion-specific ROS scavenger Mito-TEMPO (10 μM) only
partially canceled the reduction in cell viability induced by CN-A
plus PEITC (Fig. 8C). These results
suggest that ROS generation is a cause of CN-A plus PEITC-induced
cell death and that mitochondrial ROS production is not sufficient
to achieve the maximal cell death activity induced by the combined
treatment with CN-A and PEITC.
Combined treatment with CN-A and PEITC
activates ferroptosis in PANC-1 cells
Since it was recently reported that iron-dependent
ROS production in Ras-transformed cells activates programmed
necrosis in the form of ferroptosis (17), we examined the effects of the
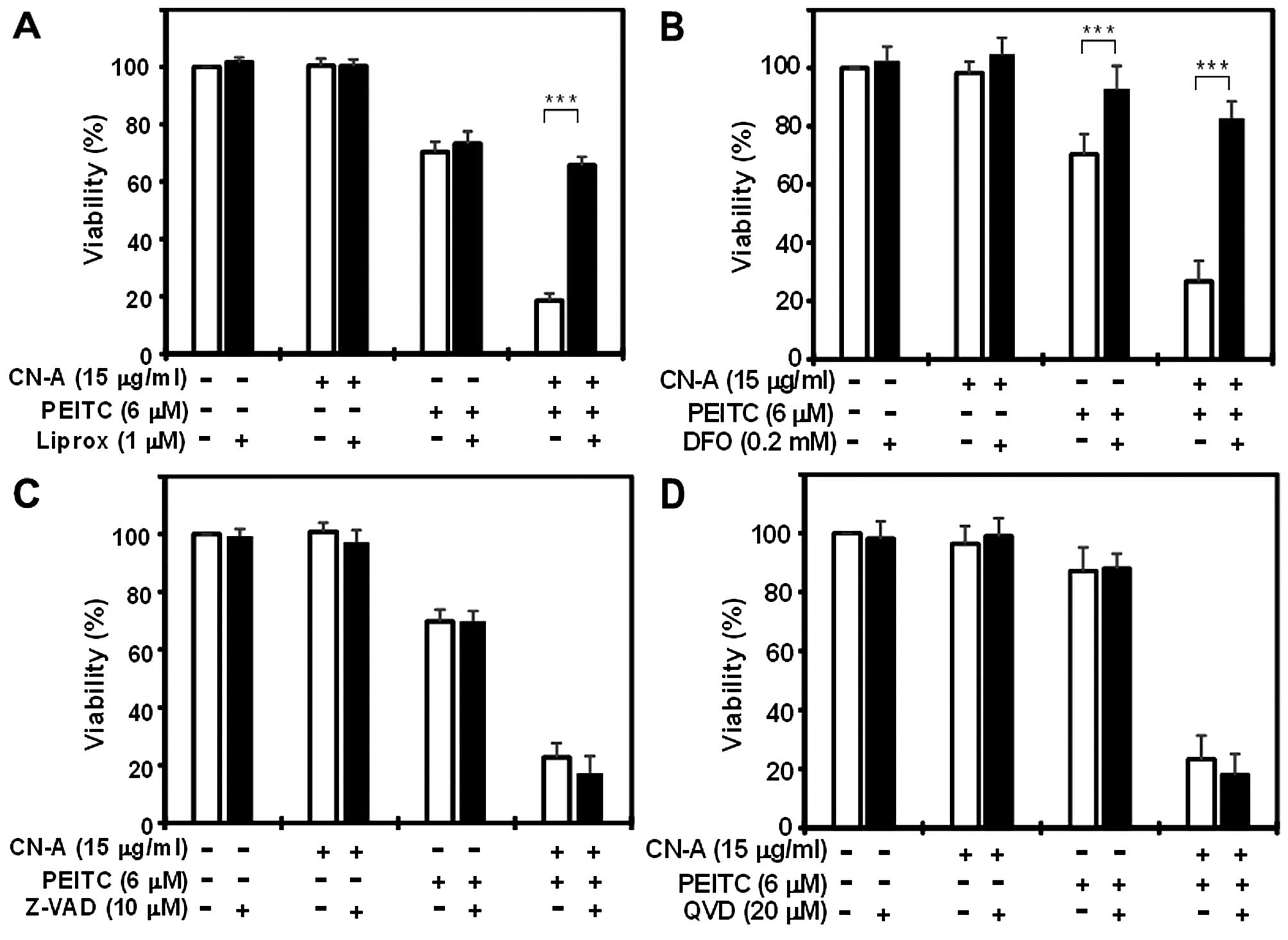
ferroptosis inhibitor Ferr-1 (17).
Ferr-1 (1.5 μM) almost completely prevented cell death
induced by CN-A plus PEITC (Fig.
9A). In contrast, it did not prevent cell death induced by
higher doses (up to 20 μM) of PEITC alone (Fig. 9B). Another potent ferroptosis
inhibitor liproxstatin (Liprox, 1 μM) (29) also protected PANC-1 cells from CN-A
plus PEITC-induced cell death (Fig.
10A). The co-addition of the lysosomal iron chelator
deferoxamine mesylate (DFO; 0.2 mM) (17) fully blocked cell death induced by
CN-A plus PEITC, demonstrating the iron dependency of cell death in
the PANC-1 cells (Fig. 10B).
Furthermore, increasing lysosomal-free iron levels by co-treatment
with iron-saturated, diferric holotransferrin (20 μg/ml)
further increased PANC-1 cell death induced by the combined
treatment (data not shown). In contrast, inhibitors of other forms
of cell death, including apoptosis [Z-VAD-FMK (Z-VAD), 10 μM
and Q-VD-OPH (QVD) (30), 20
μM] (Fig. 10C and D) and
necroptosis [Nec-1s (31), 20
μM] (Fig. 11A), failed to
suppress CN-A plus PEITC-induced cell death. The autophagy
inhibitors (3-MA, 5 mM and CQ, 20 μM) (32) partially prevented cell death
(Fig. 11B and C). Taken together,
these results suggest that synergistic cell death induced by the
combined treatment with CN-A and PEITC is mainly due to the
induction of ferroptosis.
Discussion
PEITC was previously shown to not only prevent the
initiation phase of carcinogenesis, but also to inhibit the
progression of tumorigenesis. PEITC targets multiple proteins in
order to suppress various cancer-promoting mechanisms such as cell
proliferation, progression and metastasis (9). Several mechanisms have been proposed
for the anticancer effects of PEITC (12,33–35).
PEITC is a well-known ROS inducer in cancer cells without any
potential adverse effects on normal cells (14,36).
PEITC has also been shown to activate the generation of ROS,
resulting in the downstream activation of apoptotic pathways
(13,14). In the present study, we demonstrated
that a combined treatment with CN-A and PEITC activated a form of
cell death that is distinct from canonical, caspase-mediated
apoptosis in pancreatic cancer cells. We initially reported that
PEITC in combination with CN-A activates an iron- and ROS-dependent
form of programmed necrosis, known as ferroptosis (17,18),
whereas PEITC or CN-A alone cannot induce ROS-ferroptosis
pathway-mediated pancreatic cancer cell death.
We recently found that CN-A significantly
potentiated the arsenic trioxide-induced inhibition of cell growth
in human breast cancer cells and that pretreatment with NAC
partially rescued the growth inhibition induced by the combination
of CN-A plus arsenic trioxide. The combination of CN-A plus arsenic
trioxide also synergistically increased the expression of cleaved
caspase-7 in human breast cancer MCF-7 cells and markedly inhibited
the expression of survivin (24).
These results suggest that oxidative responses are important events
in the corporative inhibition of the growth of cancer cells induced
by CN-A and arsenic trioxide and also that the combined treatment
with CN-A and arsenic trioxide induces apoptosis in cancer cells.
We speculated that the combined treatment with CN-A and ROS
inducers induces effective anti-tumor activity against solid
tumors. As described above, since PEITC is a well-known ROS inducer
in cancer cells without any potential adverse effects on normal
cells (14,36), we determined whether the combined
treatment with CN-A and PEITC exhibits effective anticancer
activity in pancreatic cancer cells. As expected, the combined
treatment with CN-A and PEITC synergistically inhibited the growth
of pancreatic cancer cells in a liquid culture and semisolid
culture (Figs. 1 and 3). In contrast to the combined treatment
with CN-A and arsenic trioxide, CN-A plus PEITC induced robust cell
death within 1 day (Figs. 4,
6 and 8) at a dose at which CN-A or PEITC alone
failed to induce prominent cell death. The combined treatment with
CN-A and PEITC synergistically increased ROS levels. This robust
cell death induced by CN-A plus PEITC was completely rescued by ROS
scavengers (NAC and trolox) (Fig. 8A
and B). However, pancaspase inhibitors (Z-VAD and QVD) failed
to rescue CN-A plus PEITC-induced cell death (Fig. 10C and D). We considered the
possibility that CN-A plus PEITC-induced cell death was not due to
apoptosis. Additionally, necroptosis was excluded since
necrostatin-1s did not rescue cell death induced by the combined
treatment (Fig. 11A). We then
explored the possibility of other forms of cell death. Our results
were consistent with the recently discovered form of cell death,
̔ferroptosis̓ (17,18). Ferroptotic cell death is
characterized by the loss of redox homeostasis, enhanced lipid
peroxidation, and inhibition by the small molecule ferrostatin-1
(Ferr-1) or iron chelator deferoxamine. Ferroptosis inhibitors
(ferrostatin-1 and liproxstatin) and deferoxamine almost completely
rescued CN-A plus PEITC-induced cell death (Figs. 9A, 10A
and B). Furthermore, iron-saturated, diferric holotransferrin,
which increases lysosomal-free iron levels, further enhanced cell
death induced by CN-A plus PEITC (data not shown). Autophagy
inhibitors (3-methyladenine and chloroquine) partially prevented
combined treatment-induced cell death (Fig. 11B and C). Thus, we presently
consider the combined treatment with CN-A and PEITC-induced cell
death of PANC-1 cells to be mainly due to ferroptosis rather than
apoptosis and necroptosis.
We previously reported a synergistic interaction
between CN-A and rapamycin in the induction of growth arrest in
mammary tumor cells (26,27,37)
and also observed a similar synergistic interaction between
ISIR-005 (a synthetic CN-A-derivative), but not FC-J (a
CN-A-related natural product), and rapamycin in the induction of
growth arrest in mammary tumor cells (27). We recently found a synergistic
interaction between CN-A and arsenic trioxide in the induction of
apoptosis in mammary tumor cells, and also observed a similar
synergistic interaction between ISIR-005, but not FC-J, and arsenic
trioxide in the induction of apoptosis in mammary tumor cells
(24). ISIR-042 (another active
synthetic CN-A-derivative) plus some antitumor agents also induced
synergistic effects to suppress the proliferation of solid tumor
cells including pancreatic cancer cells (28). Although we expected ISIR-005 and
ISIR-042 to also synergistically interact with PEITC and induce
ferroptosis, similar to CN-A, ISIR-005 plus PEITC or ISIR-042 plus
PEITC failed to induce synergistic cell death (Fig. 6). These results suggest that PEITC
specifically interacts with CN-A, but not the other CN-A
derivatives and induces ferroptotic cell death in pancreatic cancer
cells. Since only CN-A among these CN-A and its derivatives
contains an epoxide-ring, we determined whether PEITC and an agent
that contains epoxide ring(s) have the ability to induce
synergistic ferroptotic cell death in PANC-1 cells. We used
triptolide, a diterpene triepoxide extracted from the Chinese herb
Tripterygium wilfordii with potent anticancer activity
against pancreatic cancer (38), as
an epoxide-containing agent. Triptolide and PEITC only additively
induced cell death (data not shown). Furthermore, the additive cell
death induced by triptolide plus PEITC was not rescued by the
ferroptosis inhibitor ferrostain-1 (data not shown). These results
suggest that agents containing epoxide do not always
synergistically interact with PEITC to induce ferroptotic cell
death, and that CN-A and PEITC specifically interact and induce
ferroptotic cell death. The mechanisms underlying the interaction
between CN-A and PEITC have not yet been elucidated in detail.
In conclusion, we herein demonstrated that CN-A and
PEITC cooperatively suppressed growth in pancreatic cells. Our
results suggest that CN-A and PEITC induced ferroptotic cell death
in pancreatic cancer PANC-1 cells, whereas CN-A or PEITC alone did
not. These results suggest that the combination of CN-A and PEITC
may become a novel therapeutic strategy against pancreatic
cancer.
Acknowledgments
The present study was partly supported by grants
(nos. 25462007 and 16K10459) from the Ministry of Education,
Culture, Sports, Science and Technology of Japan and by a grant
from the Shimane University ̔SUIGAN̓ project.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997.PubMed/NCBI
|
|
4
|
Kroep JR, Pinedo HM, van Groeningen CJ and
Peters GJ: Experimental drugs and drug combinations in pancreatic
cancer. Ann Oncol. 10(Suppl 4): S234–S238. 1999. View Article : Google Scholar
|
|
5
|
Jakstaite A, Maziukiene A, Silkuniene G,
Kmieliute K, Gulbinas A and Dambrauskas Z: HuR mediated
post-transcriptional regulation as a new potential adjuvant
therapeutic target in chemotherapy for pancreatic cancer. World J
Gastroenterol. 21:13004–13019. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guilford JM and Pezzuto JM: Natural
products as inhibitors of carcinogenesis. Expert Opin Investig
Drugs. 17:1341–1352. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Higdon JV, Delage B, Williams DE and
Dashwood RH: Cruciferous vegetables and human cancer risk:
Epidemiologic evidence and mechanistic basis. Pharmacol Res.
55:224–236. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang L, Zirpoli GR, Guru K, Moysich KB,
Zhang Y, Ambrosone CB and McCann SE: Intake of cruciferous
vegetables modifies bladder cancer survival. Cancer Epidemiol
Biomarkers Prev. 19:1806–1811. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gupta P, Wright SE, Kim SH and Srivastava
SK: Phenethyl isothiocyanate: A comprehensive review of anti-cancer
mechanisms. Biochim Biophys Acta. 1846:405–424. 2014.PubMed/NCBI
|
|
10
|
Satyan KS, Swamy N, Dizon DS, Singh R,
Granai CO and Brard L: Phenethyl isothiocyanate (PEITC) inhibits
growth of ovarian cancer cells by inducing apoptosis: Role of
caspase and MAPK activation. Gynecol Oncol. 103:261–270. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu X, Zhou QH and Xu K: Are
isothiocyanates potential anticancer drugs? Acta Pharmacol Sin.
30:501–512. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hong YH, Uddin MH, Jo U, Kim B, Song J,
Suh DH, Kim HS and Song YS: ROS accumulation by PEITC selectively
kills ovarian cancer cells via UPR-mediated apoptosis. Front Oncol.
5:1672015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Trachootham D, Zhou Y, Zhang H, Demizu Y,
Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, et
al: Selective killing of oncogenically transformed cells through a
ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer
Cell. 10:241–252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiao D, Powolny AA, Moura MB, Kelley EE,
Bommareddy A, Kim SH, Hahm ER, Normolle D, Van Houten B and Singh
SV: Phenethyl isothiocyanate inhibits oxidative phosphorylation to
trigger reactive oxygen species-mediated death of human prostate
cancer cells. J Biol Chem. 285:26558–26569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shaw AT, Winslow MM, Magendantz M, Ouyang
C, Dowdle J, Subramanian A, Lewis TA, Maglathin RL, Tolliday N and
Jacks T: Selective killing of K-ras mutant cancer cells by small
molecule inducers of oxidative stress. Proc Natl Acad Sci USA.
108:8773–8778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang WS and Stockwell BR: Synthetic lethal
screening identifies compounds activating iron-dependent,
nonapoptotic cell death in oncogenic-RAS-harboring cancer cells.
Chem Biol. 15:234–245. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar
|
|
19
|
Sassa T, Tojyo T and Munakata K: Isolation
of a new plant growth substance with cytokinin-like activity.
Nature. 227:3791970. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Asahi K, Honma Y, Hazeki K, Sassa T,
Kubohara Y, Sakurai A and Takahashi N: Cotylenin A, a plant-growth
regulator, induces the differentiation in murine and human myeloid
leukemia cells. Biochem Biophys Res Commun. 238:758–763. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yamamoto-Yamaguchi Y, Yamada K, Ishii Y,
Asahi KI, Tomoyasu S and Honma Y: Induction of the monocytic
differentiation of myeloid leukaemia cells by cotylenin A, a plant
growth regulator. Br J Haematol. 112:697–705. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamada K, Honma Y, Asahi KI, Sassa T, Hino
KI and Tomoyasu S: Differentiation of human acute myeloid leukaemia
cells in primary culture in response to cotylenin A, a plant growth
regulator. Br J Haematol. 114:814–821. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Honma Y: Cotylenin A - a plant growth
regulator as a differentiation-inducing agent against myeloid
leukemia. Leuk Lymphoma. 43:1169–1178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kasukabe T, Okabe-Kado J, Kato N, Honma Y
and Kumakura S: Cotylenin A and arsenic trioxide cooperatively
suppress cell proliferation and cell invasion activity in human
breast cancer cells. Int J Oncol. 46:841–848. 2015.
|
|
25
|
Kawakami K, Hattori M, Inoue T, Maruyama
Y, Ohkanda J, Kato N, Tongu M, Yamada T, Akimoto M, Takenaga K, et
al: A novel fusicoccin derivative preferentially targets hypoxic
tumor cells and inhibits tumor growth in xenografts. Anticancer
Agents Med Chem. 12:791–800. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kasukabe T, Okabe-Kado J, Kato N, Sassa T
and Honma Y: Effects of combined treatment with rapamycin and
cotylenin A, a novel differentiation-inducing agent, on human
breast carcinoma MCF-7 cells and xenografts. Breast Cancer Res.
7:R1097–R1110. 2005. View Article : Google Scholar
|
|
27
|
Kasukabe T, Okabe-Kado J, Haranosono Y,
Kato N and Honma Y: Inhibition of rapamycin-induced Akt
phosphorylation by cotylenin A correlates with their synergistic
growth inhibition of cancer cells. Int J Oncol. 42:767–775.
2013.
|
|
28
|
Miyake T, Honma Y, Urano T, Kato N and
Suzumiya J: Combined treatment with tamoxifen and a fusicoccin
derivative (ISIR-042) to overcome resistance to therapy and to
enhance the antitumor activity of 5-fluorouracil and gemcitabine in
pancreatic cancer cells. Int J Oncol. 47:315–324. 2015.PubMed/NCBI
|
|
29
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xargay-Torrent S, López-Guerra M,
Montraveta A, Saborit-Villarroya I, Rosich L, Navarro A,
Pérez-Galán P, Roué G, Campo E and Colomer D: Sorafenib inhibits
cell migration and stroma-mediated bortezomib resistance by
interfering B-cell receptor signaling and protein translation in
mantle cell lymphoma. Clin Cancer Res. 19:586–597. 2013. View Article : Google Scholar
|
|
31
|
Takahashi N, Duprez L, Grootjans S,
Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van
Hauwermeiren F, Libert C, et al: Necrostatin-1 analogues: Critical
issues on the specificity, activity and in vivo use in experimental
disease models. Cell Death Dis. 3:e4372012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Carew JS, Nawrocki ST, Giles FJ and
Cleveland JL: Targeting autophagy: A novel anticancer strategy with
therapeutic implications for imatinib resistance. Biologics.
2:201–204. 2008.PubMed/NCBI
|
|
33
|
Keum YS, Jeong WS and Kong AN:
Chemoprevention by isothiocyanates and their underlying molecular
signaling mechanisms. Mutat Res. 555:191–202. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen YR, Wang W, Kong AN and Tan TH:
Molecular mechanisms of c-Jun N-terminal kinase-mediated apoptosis
induced by anticarcinogenic isothiocyanates. J Biol Chem.
273:1769–1775. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang YM, Conaway CC, Chiao JW, Wang CX,
Amin S, Whysner J, Dai W, Reinhardt J and Chung FL: Inhibition of
benzo(a)pyrene-induced lung tumorigenesis in A/J mice by dietary
N-acetylcysteine conjugates of benzyl and phenethyl isothiocyanates
during the postinitiation phase is associated with activation of
mitogen-activated protein kinases and p53 activity and induction of
apoptosis. Cancer Res. 62:2–7. 2002.PubMed/NCBI
|
|
36
|
Jutooru I, Guthrie AS, Chadalapaka G,
Pathi S, Kim K, Burghardt R, Jin UH and Safe S: Mechanism of action
of phenethylisothiocyanate and other reactive oxygen
species-inducing anticancer agents. Mol Cell Biol. 34:2382–2395.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kasukabe T, Okabe-Kado J and Honma Y:
Cotylenin A, a new differentiation inducer, and rapamycin
cooperatively inhibit growth of cancer cells through induction of
cyclin G2. Cancer Sci. 99:1693–1698. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mujumdar N, Mackenzie TN, Dudeja V, Chugh
R, Antonoff MB, Borja-Cacho D, Sangwan V, Dawra R, Vickers SM and
Saluja AK: Triptolide induces cell death in pancreatic cancer cells
by apoptotic and autophagic pathways. Gastroenterology.
139:598–608. 2010. View Article : Google Scholar : PubMed/NCBI
|