Introduction
Endometrial carcinoma is the third most common
gynecological cancer, but its frequency has increased steadily in
the last three decades (1). Most
women who present with early disease are treated with surgery
and/or radiotherapy, giving 5-year survival rates of >70%.
Approximately one-fourth of those patients go onto develop a
recurrence. Although the exact cause of endometrial carcinoma
remains unknown, the argument that estradiol somehowis implicated
in this carcinoma is becoming increasingly more difficult to
refute. On the other hand, the need of molecular-target therapy has
increased, especially for recurrent cases. Thus, a better
understanding is necessary of the molecular pathways of endometrial
carcinogenesis.
Estradiol-induced proliferation for mammary and
uterine epithelial cells is primarily mediated by estrogen receptor
(ER), which belongs to a steroid receptor superfamily/thyroid
hormone superfamily of transcription factors. This is the
classical, or genomic, pathway of the estrogen effect (2). Non-genomic estrogenic effects have
been described in a few mammalian tissues and cell lines. For
example, Sudhagar et al (3)
demonstrated that estradio activated the Akt signaling pathway in a
non-genomic manner.
The serine/threonine kinase (Akt), also known as
protein kinase B (PKB), is the cellular homologue of the viral
oncogene v-Akt. It functions as a downstream regulator of PI3K
signaling, activated by multiple growth factors. It has been
demonstrated that a subunit of phosphatidylinositol 3-kinase
(PI3-K) including the regulatory and catalytic play a role in NF-κB
activation by the tyrosine phosphorylation-dependent pathway
(4). In addition to its role in
inflammation and the suppression of apoptosis, cellular survival,
transformation and oncogenesis, the transcription factor NF-κB
plays an important role in control of the expression of many
related genes (5).
Evidence exists suggesting that estrogen-induced
angiogenesis appears to occur via induction of various angiogenic
factors such as vascular endothelial growth factor (VEGF) and basic
fibroblast growth factor (bFGF) (6,7).
However, the molecular mechanisms of how estradiol induces the
expression of these angiogenic factors are poorly understood.
Furthermore, estradiol enhances angiogenesis through a pathway
involving platelet-activating factor-mediated NF-κB activation. In
endometrial cancer cells, regulation of COX-2 protein expression by
Akt is mediated through the NF-κB/IκB pathway (8). These studies led to the hypothesis
that Akt via NF-κB plays a role in estradiol-induced angiogenesis.
The results from experiments designed to test this hypothesis
demonstrated that induction of angiogenesis occurs by increasing
the expression of several key angiogenic factors through
AKT-dependent NF-κB activation.
Materials and methods
Ethics statement
The present study was approved by the Animal Care
and Use Committee of Guangxi Medical University. The animal
research was carried out with strict adherence to the accepted
standards for the Care and Use of Laboratory Animals of China. The
protocol was approved by the Committee on the Ethics of Animal
Experiments of Affiliated Tumor Hospital of Guangxi Medical
University [ethical approval reference: CS2012(28)]. All efforts were made to minimize
animal suffering.
Cell culture and reagents
The human endometrial cancer Ishikawa and HEC-1A
cell line, positive and negative with estrogen receptor,
respectively, were generous gifts from Dr Wei Lihui, Gynecological
Oncology Center of Peking University People's Hospital. Cells were
maintained at 37°C under 5% CO2 in Dulbecco's modified
Eagle's medium (DMEM; HyClone Laboratories, Inc., Logan, UT, USA)
supplemented with 10% fetal bovine serum (FBS; HyClone
Laboratories), 100 U/ml penicillin and 100 mg/ml streptomycin. The
medium was changed every three days.
Proliferation assays
The logarithmic phase cells were digested,
centrifuged and labeled with concentration of 5 µM CFSE
(Molecular Probes, Eugene OR, USA), then 1×105 cells
were seeded per well in 6-well plates. When the cells attached, the
supernatant was changed with 1×10−6 mol/l estradiol or
vehicle to different 3-wells, after 30 min, the effect of estradiol
or vehicle was terminated through changing the supernatant to DMEM
with 10% FBS, the cells were cultured for 24, 48, 72 and 96 h.
Then, flow cytometry (BD FACSCanto™; BD Biosciences, Piscataway,
NJ, USA) was used to detect the proliferation of cells. The results
were analyzed by using ModFit LT 3.0 software.
Clone formation assay
Cells were seeded in 6-well plates (500 cells/well),
when adhered, they were treated with 1×10−6 mol/l
estradiol, or DMEM as control for 30 min. The medium of 6-wells
were all changed to DMEM with 10% FBS to terminate the effect of
estradiol or control. After 7 days, cells were fixed by 95%
methanol for 15 min and stained with H&E. Clones containing ≥50
cells were counted under the low power microscope (×10). The clone
formation rate was calculated using the following formula: clone
formation rate (%) = (number of clones/number of seeded cells) ×
100%.
Cell cycle analysis
Ishikawa or HEC-1A (1×106) cells were
seeded in a 6-well plate respectively, when the cells attached, the
supernatant was discarded, and 1.5 ml FBS-free DMEM was added for
24 h to induce cell cycle in G0 phase. Then, 1.5 ml DMEM was added
with 1×10−6 mol/l E2 to 3-wells as E2 group, or DMEM
only as control. After 30 min, the medium of 6-wells were all
changed to DMEM with 10% FBS to terminate the effect of estradiol
or control. The cells were grown continuously at 37°C in a humid
chamber with 5% CO2. After 24 h, cells were treated
according to the instructions of cycle test plus DNA reagent kit
(BD Biosciences) and analyzed by flow cytometry (FCM; BD
FACSCanto). The results were analyzed by ModFit LT 3.0
software.
Transwell migration and Matrigel invasion
assay
The cell migration/invasion experiment was performed
in Transwell mini-chamber (Milipore, Billerica, MA, USA). For
Transwell invasion assay, the membrane of the upper chamber was
pre-coated with Matrigel at a 1:3 dilution (BD Biosciences).
After treated with 1×10−6 mol/l
estradiol, or DMEM as control for 30 min, 1×105 cells
were seeded in the upper wells filled with serum-free medium, the
lower chambers were filled with complete medium supplemented with
20% FBS to induce cell migration. After the cells were incubated at
37°C for 24 h, the migrated/invasived cells through the membrane to
the lower side were fixed, stained with crystal violet and
photographed. Five random fields per insert were imaged at ×200
magnification. Migrated/invasived cells in the images were
counted.
Semi-quantitative RT-PCR and ELISA for
VEGF and bFGF expression
After adherence, cells were cultured in DMEM without
FBS for 24 h till ~70–80% confluence. After being pretreated with
25×10−6 mol/l AKT inhibitor
1L-6-Hydroxymethyl-chiro-inosito12-[(R)-2-0-methyl-3-0-octadecylcarbonate]
(Alexis Co., Farmers Branch, TX, USA) (AKT inhibitor group, AKTI),
or 1×10−6 mol/l ICI 182,780 (estrogen receptor
antagonist; Tocris Bioscience Co., Bristol, UK) (ER inhibitor
group, EI), or 100 µmol/l NF-κB inhibitor PDTC; Sigma, St.
Louis, MO, USA) (NF-κB inhibitor group, PDTC) for 60 min, cells
were stimulated with 1×10−6 mol/l β-Estradiol-water
soluble (Sigma) (E2 group) for 30 min while control group was
treated with FBS-free DMEM.
Total RNA was extracted from the cells using TRIzol
reagent (Fermentas, Burlington, ON, Canada) according to the
manufacturer's instructions. The total RNA was reverse transcribed
using RevertAid™ First Strand cDNA Synthesis kit (Fermentas). The
sequences for the forward and reverse primers for human VEGF are
5′-GAATCATCACGAAGTGGTGAAGT-3′ and 5′-GCACACAGGATGGCTTGAAG-3,
respectively. The sequences for the forward and reverse primers for
human bFGF are 5′-TATTTCTTTGGCTGCTACTTG-3′ and
5′-TCCAGCATTTCGGTGTTG-3′, respectively. The sequences for the
forward and reverse primers for human GAPDH are
5′-GAAGGTGAAGGTCGGAGT-3′ and 5′-GAAGATGGTGATGGGATTTC-3′,
respectively. All the sequences were synthesized by Invitrogen
(Carlsbad, CA, USA). FastStart Universal SYBR-Green Master (Rox)
was from Roche (Indianapolis, IN, USA). The real-time PCR protocol
included 94°C for 3 min; 40 cycles of 94°C for 30 sec, 55°C for 30
sec and 72°C for 30 sec, final extension were 72°C for 10 min.
The quantitative determination of VEGF and bFGF in
culture supernatant was performed by ELISA kit (Shanghai ExCell
Biology, Inc., Shanghai, China) according to the manufacturer's
instructions. The absorbance of 450 nm was measured and the
expression of VEGF and bFGF evaluated in the cell culture
supernatant.
NF-κB activation
Cells were treated with different concentrations of
estradiol (10−4, 10−6, 10−8 and
10−10 mol/l) for 30 min, or 1×10−6 mol/l
estradiol at different time-points (15 and 30 min, 1 and 2 h), or
25×10−6 mol/l AKT inhibitor for 60 min following
incubation with 1×10−6 mol/l estradiol for 30 min.
Harvested cells were washed, centrifuged at 500 rpm for 5 min and
mixed with PBS/phosphatase inhibitor, then nucleoprotein was
prepare using nuclear extract kit (Active Motif Co., Carlsbad, CA,
USA). Quantitative NF-κB DNA-binding p65 was determined using
ELISA-based TransAM™ assays (Active Motif) in accordance with the
manufacturer's instructions. The kit contain a 96-well plate
pre-immobilized oligonucleotide containing the NF-κB consensus site
5′-GGGACTTTCC-3′. Activated NF-κB nuclear extraction is first bound
to the attached oligonucleotide. The primary antibody was used to
detect an epitope on p65 recognized by NF-κB. An enzyme
(HRP)-linked secondary antibody was quantified by spectrophotometry
(450 nm absorbance) to evaluated the NF-κB activation.
Western blot analysis
Cells were lysed using lysis buffer with protease
inhibitor, after 30-min incubation on ice, centrifuged with 12,000
rpm for 10 min, the supernatant was collected. The protein
concentration was determined by BCA (bicinchoninic acid assay;
Fermentas). Twenty-five micrograms protein per each lane was
separated electrophoretically by 10% SDS-PAGE gel. The gel was
transferred to PVDF membrane, then the membrane was blocked in 5%
non-fat milk in PBS for 2 h at room temperature followed by washes
with 1X TBST. The membranes were incubated with primary antibodies
against phospho-Akt (ser 473), AKT or GAPDH (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. The membrane
was washed three times with TBS-T for 20 min and then incubated for
2 h with peroxidase-labeled anti-rabbit IgG (KPL, Gaithersburg, MD,
USA) at room temperature in 5% non-fat milk in TBST, subsequently
washed with TBST three times for 20 min. The signal was developed
by addition of enhanced chemiluminescence solution (Bio-Rad
Laboratories, Hercules, CA, USA). The proteins were detected with
infrared imaging system. Band intensity was quantified using
infrared imaging system. Akt and phospho-Akt (p-Akt) band
intensities were normalized to those of GAPDH.
Animal care and experiments
Specific pathogen-free female BALB/C mice were
obtained from the Animal Experiment Centre of Guangxi Medical
University and maintained in our animal facility. All mice were
used at 3–4 weeks of age. Subcutaneous xenograft tumors in BALB/c
mice were established by the inoculation of a 0.2 ml cell
suspension containing 1×107 Ishikawa or HEC-1A cells in
each of the 15 mice. When xenograft tumors reached 1
cm3, mice were assigned randomly to one of five
treatment regimens once every three days, seven times: i) 25 ml/kg
estradiol (E2); ii) 45 mg/kg estrogen inhibitor ICI 182,780 (EI);
iii) 25 ml/kg AKT inhibitor (AKTI); iv) 50 mg/kg NF-κB inhibitor
PDTC (PDTC); v) vehicle (control) were injected into the abdomen,
respectively. Each treatment group consisted of three mice. The day
following last inoculation, mice were weighed and sacrificed. Tumor
volumes (cm3) were calculated by the formula: [(major
axis) × (minor axis)2/2]. After the treatment, the
tumors were removed and analyzed by immunohistochemistry.
Immunohistochemistry
VEGF, phospho-AKT, AKT and GAPDH antibody (rabbit
IgG) were purchased from Santa Cruz Biotechnology. bFGF and
NF-κB/p65 antibody (rabbit IgG) were from Cell Signaling Technology
(Danvers, MA, USA). Normal rabbit IgG as negative control antibody
were from Cell Signaling Technology. DyLight 680 goat anti-rabbit
IgG (H+L) was from KPL.
Tissues were fixed in 10% formalin buffer, embedded
in paraffin, and sectioned at 4 µm for consecutive glass
slides which were incubated with normal rabbit IgG (1:200, as
negative control), or rabbit polyclonal anti-human VEGF antibody
(1:120), or anti-human bFGF antibody (1:120), or p-Akt (Ser473)
antibody (1:200), or NF-κB/p65 antibody (1:200) for 1 h at room
temperature. Slides were rinsed in PBS and incubated with
anti-rabbit secondary antibody. The subsequent steps were carried
out using the SP (streptavidin-peroxidase) kit (Zymed Laboratories,
Inc., South San Francisco, CA, USA) according to the manufacturer's
instruction. After DAB solution was applied, slides were
counterstained with hematoxylin and then evaluated under a light
microscope. Cytoplasmic staining intensity, and the proportion of
positive tumor cells were recorded and the staining intensity (0–3)
and the area of positive staining (0, no staining; 1+, <30%; 2+,
30–60%; 3+, >60%) were calculated.
Statistical analysis
All statistical analyses were performed with SPSS
statistical software (version 19.0). The data are represented as
the mean ± standard deviation (SD). Analysis of variance was used
to determine P-value between mean measurements. P-value of <0.05
was deemed to be statistically significant. All experiments were
conducted in triplicate and repeated two or more times.
Results
Effect of estradiol on cell biological
behavior
To determine the role of estradiol on cell
proliferation, Ishikawa and HEC-1A cells were treated with or
without 1×10−6 mol/l estradiol for 24, 48, 72 and 96 h.
Cell proliferation of E2 groups was significantly faster than that
in control group in different time-points. The results showed that
estradiol can promote Ishikawa and HEC-1A cell division and
proliferation (Fig. 1).
To evaluate the impact of estradiol on Ishikawa and
HEC-1A cell growth, clone-forming experiment was developed. A total
of 500 cells were seeded and treated with 1×10−6 mol/l
estradiol or with DMEM as control for 30 min. After 7 days, the
clone formation rate in E2 group was significantly higher than that
in control group (Fig. 2C).
The migration experiment was performed to verify the
effect on motility of Ishikawa and HEC-1A cells. The cells that
managed to traverse the filter to the lower chamber were counted
under magnification. The migrating cells in E2 group was
significantly increased compared to that in control group (Fig. 2A and D). The invasion capacity of
Ishikawa cells was evaluated in Matrigel invasion assays. The
number of cells that invaded through the Matrigel and membrane was
determined. The invading cells in E2 group was significantly
increased compared to the control group (Fig. 2A and D). These results suggest that
estradiol may impact tumor metastatic ability in endometrial cancer
cells.
To further observe the estradiol function on the
cell cycle, 1×106 cycle synchronized cells, after serum
starvation, were seeded in 6-well plates and treated with or
without 1×10−6 mol/l estradiol for 30 min, G0/G1
percentage of E2 groups were significantly lower than that in
control groups, percentage of S phase cells in E2 groups were
significantly increased compared to that in control groups
(Fig. 2B and E), suggesting that
estradiol might stimulate cell proliferation.
Estradiol induced mRNA expression of VEGF
and bFGF via Akt induced NF-κB pathway in the endometrial cancer
Ishikawa cells
To understand the function of estradiol and
angiogenesis in endometrial cancer, we first investigated whether
estradiol induced mRNA expression and protein synthesis of the VEGF
and bFGF in Ishikawa and HEC-1A cells through real-time
semi-quantitative RT-PCR and ELISA for VEGF and bFGF expression.
The effect of VEGF and bFGF level increased in Ishikawa cells with
estradiol treatment could be blocked by estradiol inhibitor ICI,
AKT inhibitor, or NF-κB inhibitor PDTC. However, the effect of VEGF
and bFGF level increased in HEC-1A cells with estradiol treatment
could be blocked by AKT inhibitor, NF-κB inhibitor PDTC, but not by
the estradiol inhibitor ICI (Fig.
3), suggesting a role of Akt and NF-κB in estradiol-induced
angiogenic factor synthesis.
 | Figure 3Involvement of AKT via NF-κB in the
induction of VEGF and bFGF production by estradiol. (A) VEGF and
bFGF mRNA expression in Ishikawa cells pretreated with estradiol
receptor inhibitor ICI, AKT inhibitor, NF-κB inhibitor PDTC,
respectively, at indicated concentrations before 1×10−6
mol/l estrogen treatment was detected through real-time RT PCR.
VEGF and bFGF mRNA increase in Ishikawa cells with estradiol
treatment could be blocked by estradiol receptor inhibitor ICI, AKT
inhibitor, or NF-κB inhibitor PDTC. (B) VEGF and bFGF protein in
the Ishikawa cell culture supernatant was measured by ELISA. VEGF
and bFGF protein level tendency was consistent with mRNA
expression. (C) VEGF and bFGF mRNA expression in HEC-1A cells
pretreated with estradiol receptor inhibitor ICI, AKT inhibitor,
NF-κB inhibitor PDTC, respectively, at indicated concentrations
before 1×10−6 mol/l estrogen treatment was detected
through real-time RT PCR. VEGF and bFGF mRNA increased in Ishikawa
cells with estradiol treatment could be blocked by AKT inhibitor,
or NF-κB inhibitor PDTC, but not by the estradiol receptor
inhibitor ICI. (D) VEGF and bFGF protein in the HEC-1A cell culture
supernatant was measured by ELISA. VEGF and bFGF protein level
tendency was consistent with mRNA expression. Each bar represents
the mean ± SD. *P<0.01, compared with control group;
#P<0.05, compared with E2 group. |
Effects of estradiol on Akt kinase
activity
To determine whether estradiol can also activate the
Akt pathway, the ability of estradiol to induce Akt protein
expression was determined by western blot analysis. Ishikawa and
HEC-1A cells were serum starved for 24 h and treated with
1×10−6 mol/l estradiol for 30 min with or without
25×10−6 mol/l of Akt inhibitor, or 1×10−6
mol/l of anti-estradiol ICI 182,780 preincubation for 1 h. We
detected total Akt and phosphorylation Akt of Ser473 to evaluate
activated serine/threonine kinase function of Akt. The ratio of
p-Akt (at Ser473 site)/Akt/GAPDH was used as the level of Akt
activation. The results showed that Akt activation was stimulated
by estradiol. In contrast, ICI 182,780 and Akt inhibitor inhibited
estrogen-induced Akt activation in Ishikawa cells (Fig. 4A and B) while only Akt inhibitor
inhibited estrogen-induced Akt activation in HEC-1A (Fig. 4C and D).
NF-κB activation by Akt in endometrial
cancer cells
To investigate whether estradiol could regulate
NF-κB activity, we measured its activity in Ishikawa and HEC-1A
cells stimulated by varied concentrations of estradiol at 30 min.
NF-κB activity peaked at concentration of 1×10−6 mol/l
estradiol (Fig. 5A and D). After
incubation with 1×10−6 mol/l estradiol, the level of
NF-κB activity in Ishikawa cells was gradually increased at 30 min
and peaked at 60 min, showing a time-dependent manner (Fig. 5B and E). The results indicated that
estradiol could induce NF-κB activation.
 | Figure 5NF-κB activity. (A) The NF-κB
activity in Ishikawa cells after stimulated with concentration of
1×10−6 mol/l estradiol were significantly higher than
that in the control group and the other concentration groups
(10−4, 10−8 and 10−10 mol/l);
*P<0.05. (B) The NF-κB activity in Ishikawa cells
after stimulated with 1×10−6 mol/l estradiol for 30 min
and 1 h were significantly higher than that in 15 min and 2 h;
*P<0.05. (C) Ishikawa cells was incubated with
1×10−6 mol/l estradiol for 30 min (E2 group), or with
the AKT inhibitor for 1 h, following incubation with
1×10−6 mol/l estradiol for 30 min, or vehicle as
control. NF-κB activity in AKT inhibitor group was significantly
lower than that in estradiol group. *P<0.05, AKT
inhibitor group compared with E2 group. (D) The NF-κB activity in
HEC-1A cells after stimulated with concentration of
1×10−6 mol/l estradiol were significantly higher than
that in the control group and the other concentration groups
(10−4, 10−8 and 10−10 mol/l);
*P<0.05. (E) The NF-κB activity in HEC-1A cells after
stimulated with 1×10−6 mol/l estradiol for 30 min and 1
h were significantly higher than that in 15 min and 2 h;
*P<0.05. (F) HEC-1A cells was incubated with
1×10−6 mol/l estradiol for 30 min (E2 group), or with
the AKT inhibitor for 1 h, following incubation with
1×10−6 mol/l estradiol for 30 min, or vehicle as
control. NF-κB activity in AKT inhibitor group was significantly
lower than that in estradiol group. *P<0.05, AKT
inhibitor group compared with E2 group. Each bar represent the mean
± SD of 3 independent experiments. |
To further study the relationship between Akt and
NF-κB activation, Ishikawa and HEC-1A cells were serum starved for
24 h and then treated with 1×10−6 mol/l estradiol for 30
min with or without 25×10−6 mol/l of Akt inhibitor
preincubation. NF-κB activity in AKT inhibitor was significantly
weaker than that in estradiol stimulation (Fig. 5C and F). Thus, NF-κB activity
induced by estradiol was blocked by AKT inhibitor pretreatment,
confirming that estradiol induced NF-κB activation by Akt. Taken
together, these data demonstrated that estradiol can mediate the
expression of VEGF and bFGF through AKT pathway involving NF-κB
activation.
Growth of tumors in vivo
To further elucidate the mechanism by which
estradiol and Akt-mediated NF-κB pathway stimulates growth of
endometrial cancer cells, we developed Ishikawa and HEC-1A
cell-stimulated endometrial cancer model in vivo. In this
model, tumors originated from Ishikawa or HEC-1A cells treated with
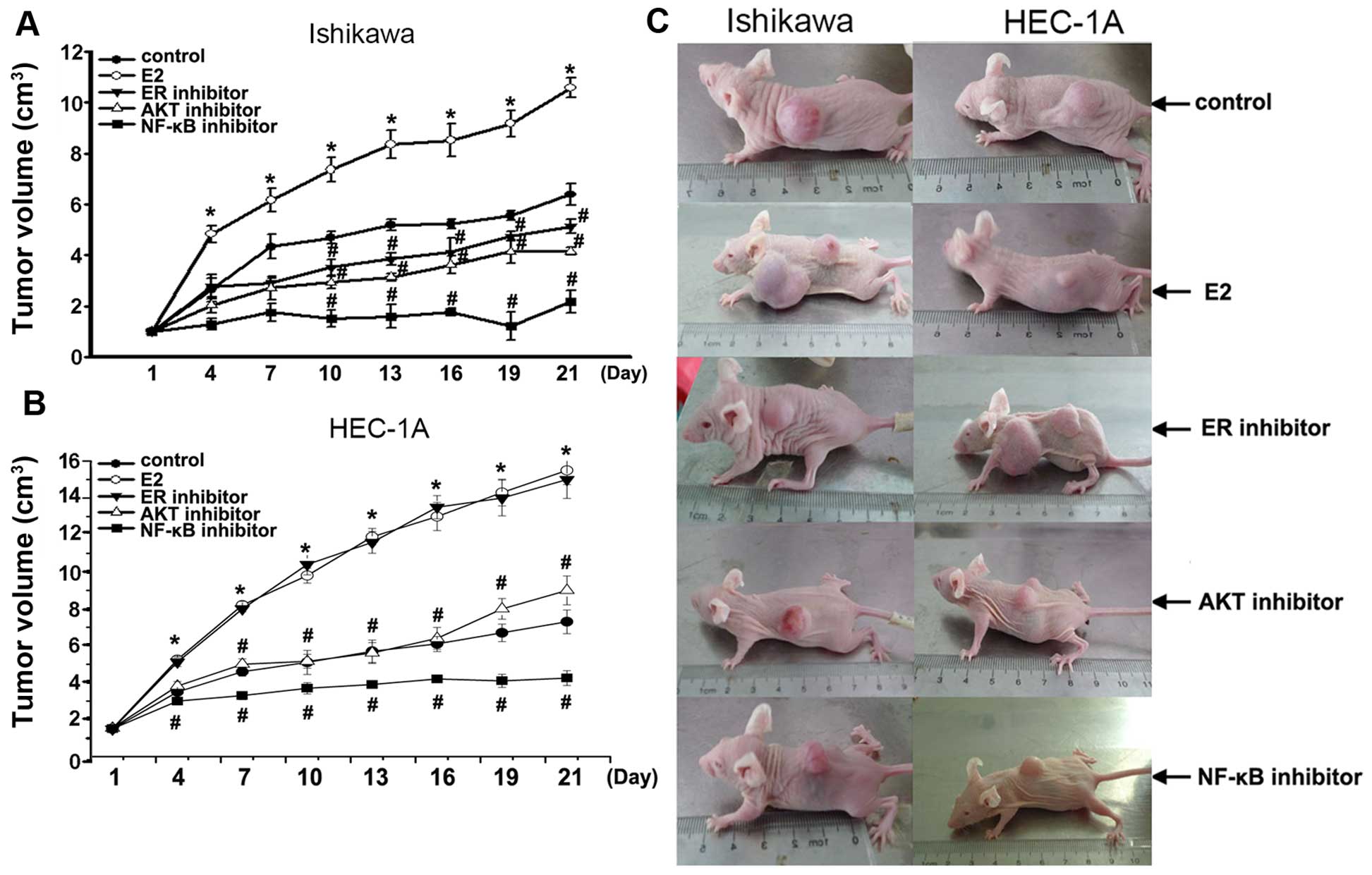
estradiol E2 grew faster than that in control (P<0.01) (Fig. 6). In Ishikawa cell model in
vivo, tumors were smaller in estrogen inhibitor ICI 182,780
alone (P<0.01), AKT inhibitor alone (P<0.01), NF-κB inhibitor
PDTC alone (P<0.01) compared with that in control. In HEC-1A
cell model in vivo, tumors were smaller in AKT inhibitor
alone (P<0.01), NF-κB inhibitor PDTC alone (P<0.01) compared
with that in control. Notably, NF-κB inhibitor PDTC particularly
blocked xenograft tumors, compared with estrogen inhibitor or AKT
inhibitor. These data showed that tumor continues to grow in
response to E2, but was resistant to its Akt-mediated NF-κB pathway
inhibitor in the model in vivo of Ishikawa or HEC-1A
cells-stimulated endometrial tumors.
We constructed a xenograft model in nude mice by
inoculation of Ishikawa or HEC-1A cells. When xenograft tumor was
developed to 1 cm3, we investigated the effect after
using E2, estrogen inhibitor ICI 182,780, AKT inhibitor, or NF-κB
inhibitor PDTC through hematoxylin and eosin stain and
immunohistochemical methods.
VEGF was detected in the cytoplasm; bFGF, Akt
protein and NF-κB protein were detected mainly in the endometrial
gland epithelium and slightly in cell membrane (Fig. 7A).
 | Figure 7Immunohistochemical experiments in
the xenograft model. (A) The a, c, e and g show strong positive
immunohistochemical staining of VEGF, bFGF, AKT and NF-κB images;
the b, d, f and h are negative staining images. (B) The histogram
of VEGF, bFGF, AKT, NF-κB strong positive expression in Ishikawa
cells. (C) The histogram of VEGF, bFGF, AKT, NF-κB strong positive
expression in HEC-1A cells. |
The percentage of strong positive expression of VEGF
and bFGF in ishikawa cell estradiol group were significant higher
than that in control group (both 83.33 vs. 50.0%). The strong
positive expression of VEGF and bFGF using estrogen inhibitor ICI
182,780, or AKT Inhibitor was 0%, estradiol group was 83.33%, there
was a significant difference (P<0.05). The strong positive
expression of VEGF, and bFGF using PDTC were significantly lower
than that in E2 group (33.33, 16.67 vs. 83.33%), (P<0.05)
(Fig. 7B). The strong positive
expression rate of Akt protein in ishikawa cell E2 group was much
higher than that in control group, EI group, Akt inhibitor group,
PDTC group (95.83% vs. 66.67, 0, 0 and 33.33%, respectively;
P<0.05 all) (Fig. 7B).
The strong positive expression rate of NF-κB protein
in Ishikawa cell E2 group was much higher than that in control
group (100 vs. 62.5%; P<0.05). The strong positive expression
rate of NF-κB protein in EI group, Akt group, or NF-κB group was
significantly lower than that in E2 group (0%, 33.33, 33.33 vs.
100%, respectively; P<0.01) (Fig.
7B).
HEC-1A cell line showed almost the same results,
except in the group of EI. That is to say, for HEC-1A cell line
negative of estrogen receptor, the strong positive expression rate
of VEGF, bFGF, AKT and NF-κB protein was not significantly
different between the groups E2 and EI.
Discussion
The actions of estradiol are essential for the
growth and survival of tumors that develop from reproductive
tissue. Estradiol is thought to exert its biological effects by at
least two mechanisms: one termed genomic and the other non-genomic
(9). The rapid, non-genomic actions
of this sex steroid are attributed to membrane action. However,
studies also found that E2 attributed to rapid, non-genomic effects
on cell membrane-initiated signaling (10). Many studies show that estradiol can
induce Akt activation of PI3K/Akt which is linked to
estradiol-induced endometrial epithelial cell proliferation
(11). Estradiol can activate Akt
rapidly in endometrial cancer while blocked by PI3K inhibitor,
indicating the estradiol may activate PI3k/Akt kinase cascade
through non-genomic action involved in the occurrence of
endometrial cancer (12). E2
activation of PI3K is required for subsequent downstream activation
of membrance-associated signaling pathway-phosphorylation of Akt.
We have observed that estradiol treatment of Ishikawa or HEC-1A
cells for 30 min rapidly induce phosphorylation of Akt (Ser473). We
propose that estrogen-mediated Akt activation is non-genomic
because production of p-Akt protein was only 30 min and not
completely depend on the estrogen receptor. On the contrary, the
blocking effect of Akt inhibitor is more significant than that of
ER inhibitor in Ishikawa cells. We deduced estradiol might activate
Akt pathway by non-genomic estrogenic effects, suggesting p-Akt is
related to occurrence of the tumor.
AKT is a serine/threonine protein kinase, also known
as protein kinase B including N-terminal pleckstrin homology (PH)
domain, middle of the kinase activity domain, C-terminal regulatory
domain (RD). Akt is activated by phospholipid binding and
activation loop phosphorylation at Thr308 by PDK1 and by
phosphorylation within the carboxy-terminus at Ser473. Whereas the
phosphorylation at Ser473, an indication of the maximal activation
of Akt appears to be dependent on PI3K (13,14).
Some studies (15–17) have indicated that Akt may form a
complex with IκB kinase (IKK) and subsequently protect cells from
apoptosis. Akt phosphorylation has been shown to activate NF-κB in
RL-95-2 and Ishikawa human endometrial cancer cell lines (8). To date, it is unclear whether diverse
estradiol mediated biological effects are via Akt mediated NF-κB
pathway. Indeed, our results demonstrated that the NF-κB activity
was induced by different concentrations of estradiol. The estradiol
induced NF-κB activity was blocked by pretreatment with the Akt
inhibitor comparing with estradiol treatment, suggesting Akt
pathway is essential for the effect of estradiol on the downstream
target NF-κB.
Akt, a major effector protein kinase in the PI3-K
signaling, is regulated by steroid hormones (18), growth factors (19) and their receptors (20). ER mediated VEGF and bFGF protein
expression were shown to be strictly dependent on Akt pathway which
was inhibited by AKT inhibitors in vivo.
High expression of NF-κB in breast and endometrial
cancer showed a possible important role of NF-κB in promoting
proliferation, suppressing apoptosis and promoting cell migration
(21–23). The activity of NF-κB is tightly
controlled by inhibitory IκB proteins that bind to NF-κB complexes
and thus sequester NF-κB in the cytoplasm. Upon appropriate
stimulation, phosphorylation of IκB family members by IKB kinase
(IKK) complex leading to NF-κB nuclear translocation and
transcriptional activation. Since Akt phosphorylation has been
shown to activate NF-κB in other systems (24), a similar sequence of events might be
involved in phosphor-Akt expressing Ishikawa and HEC-1A cells in
the present study.
Regulation of the various key angiogenic factor
synthesis such as VEGF and bFGF, are controlled via NF-κB activity
(8,25). NF-κB is an important upstream
regulator of VEGF, a major angiogenic factor that induces
endothelial cell proliferation (26), promotes tumor-induced angiogenesis.
Estradiol-induced angiogenesis appear to be dependent on angiogenic
factors such as VEGF and bFGF. In the present study, we
demonstrated that estradiol induces Akt production, which
subsequently leads to production of angiogenic factors VEGF and
bFGF in endometrial cancer cells via the activation of NF-κB, this
estrogenic effect was blocked by the estradiol inhibitor ICI, AKT
inhibitor, or the NF-κB inhibitor PDTC in estrogen
receptor-positive Ishikawa cell line and blocked by AKT inhibitor,
NF-kB inhibitor PDTC in estrogen receptor-negative HEC-1A cell
line.
To the best of our knowledge, this is the first
report demonstrated the critical role of Akt-induced NF-κB
activation in estradiol-mediated angiogenesis which promotes the
cell proliferation, clone formation while impacts tumor cell
migration and invasion. In the present study, we demonstrate that
Akt is involved in estradiol induced angiogenesis because it was
observed that the inhibition of Akt or NF-κB action decreased the
effects of estradiol. We have also shown that Akt exerts its
biological activity through NF-κB activation (4,17).
Several investigators have reported a role of NF-κB in angiogenesis
(27,28). Because Akt is a critical factor of
NF-κB in vivo as well as in vitro (29), any circumstance of release and/or
synthesis of Akt abnormally may lead to pathological angiogenesis
via an NF-κB-dependent mechanism. NF-κB inhibition by parthenolide
markedly enhances the sensitivity of resistant breast cancer tumor
cells to tamoxifen through suppression of the Akt pathway (30). Thus, in the present study, we have
demonstrated that estradiol induces NF-κB activation through Akt
induction in endometrial cells.
Angiogenesis is essential for tumor growth, invasion
and metastatic spread. High expression of VEGF and bFGF were
significantly associated with histologic grade, vascular invasion
and disease progress (31,32). Therefore, VEGF and bFGF are
considered as important angiogenic and prognostic factors. In
conclusion, our findings demonstrated that estradiol induces the
synthesis of key angiogenic factors VEGF and bFGF in endometrial
cells through Akt-mediated activation of NF-κB, suggesting the
possibility of a potential therapeutic target for human endometrial
cancers.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (no. 81160318) and the Project
of Natural Science Foundation of Guangxi (no.
2013GXNSFAA019219).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mangelsdorf DJ, Thummel C, Beato M,
Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M,
Chambon P, et al: The nuclear receptor superfamily: The second
decade. Cell. 83:835–839. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sudhagar S, Sathya S and Lakshmi BS: Rapid
non-genomic signalling by 17β-oestradiol through c-Src involves
mTOR-dependent expression of HIF-1α in breast cancer cells. Br J
Cancer. 105:953–960. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Béraud C, Henzel WJ and Baeuerle PA:
Involvement of regulatory and catalytic subunits of
phosphoinositide 3-kinase in NF-kappaB activation. Proc Natl Acad
Sci USA. 96:429–434. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang CY, Mayo MW and Baldwin AS Jr: TNF-
and cancer therapy-induced apoptosis: Potentiation by inhibition of
NF-kappaB. Science. 274:784–787. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saarinen NM, Abrahamsson A and Dabrosin C:
Estrogen-induced angiogenic factors derived from stromal and cancer
cells are differently regulated by enterolactone and genistein in
human breast cancer in vivo. Int J Cancer. 127:737–745. 2010.
View Article : Google Scholar
|
|
7
|
Heaney AP, Fernando M and Melmed S:
Functional role of estrogen in pituitary tumor pathogenesis. J Clin
Invest. 109:277–283. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
St-Germain ME, Gagnon V, Parent S and
Asselin E: Regulation of COX-2 protein expression by Akt in
endometrial cancer cells is mediated through NF-kappaB/IkappaB
pathway. Mol Cancer. 3:72004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hammes SR and Davis PJ: Overlapping
nongenomic and genomic actions of thyroid hormone and steroids.
Best Pract Res Clin Endocrinol Metab. 29:581–593. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
François CM, Wargnier R, Petit F, Goulvent
T, Rimokh R, Treilleux I, Ray-Coquard I, Zazzu V, Cohen-Tannoudji J
and Guigon CJ: 17β-estradiol inhibits spreading of metastatic cells
from granulosa cell tumors through a non-genomic mechanism
involving GPER1. Carcinogenesis. 36:564–573. 2015. View Article : Google Scholar
|
|
11
|
Zhang H, Zhao X, Liu S, Li J, Wen Z and Li
M: 17betaE2 promotes cell proliferation in endometriosis by
decreasing PTEN via NFkappaB-dependent pathway. Mol Cell
Endocrinol. 317:31–43. 2010. View Article : Google Scholar
|
|
12
|
Guzeloglu Kayisli O, Kayisli UA, Luleci G
and Arici A: In vivo and in vitro regulation of Akt activation in
human endometrial cells is estrogen dependent. Biol Reprod.
71:714–721. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chan TO and Tsichlis PN: PDK2: A complex
tail in one Akt. Sci STKE 2001. pe12001.
|
|
14
|
Scheid MP and Woodgett JR: PKB/AKT:
Functional insights from genetic models. Nat Rev Mol Cell Biol.
2:760–768. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Madrid LV, Wang CY, Guttridge DC,
Schottelius AJ, Baldwin AS Jr and Mayo MW: Akt suppresses apoptosis
by stimulating the transactivation potential of the RelA/p65
subunit of NF-kappaB. Mol Cell Biol. 20:1626–1638. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR,
Pfeffer LM and Donner DB: NF-kappaB activation by tumour necrosis
factor requires the Akt serine-threonine kinase. Nature. 401:82–85.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Romashkova JA and Makarov SS: NF-kappaB is
a target of AKT in anti-apoptotic PDGF signalling. Nature.
401:86–90. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bryant DN, Sheldahl LC, Marriott LK,
Shapiro RA and Dorsa DM: Multiple pathways transmit neuroprotective
effects of gonadal steroids. Endocrine. 29:199–207. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Frago LM, Pañeda C, Argente J and Chowen
JA: Growth hormone-releasing peptide-6 increases insulin-like
growth factor-I mRNA levels and activates Akt in RCA-6 cells as a
model of neuropeptide Y neurones. J Neuroendocrinol. 17:701–710.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanda S, Miyata Y and Kanetake H:
Fibroblast growth factor-2-mediated capillary morphogenesis of
endothelial cells requires signals via Flt-1/vascular endothelial
growth factor receptor-1: Possible involvement of c-Akt. J Biol
Chem. 279:4007–4016. 2004. View Article : Google Scholar
|
|
21
|
Biswas DK, Shi Q, Baily S, Strickland I,
Ghosh S, Pardee AB and Iglehart JD: NF-kappa B activation in human
breast cancer specimens and its role in cell proliferation and
apoptosis. Proc Natl Acad Sci USA. 101:10137–10142. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou Y, Eppenberger-Castori S, Marx C, Yau
C, Scott GK, Eppenberger U and Benz CC: Activation of nuclear
factor-kappaB (NFkappaB) identifies a high-risk subset of
hormone-dependent breast cancers. Int J Biochem Cell Biol.
37:1130–1144. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mizumoto Y, Kyo S, Kiyono T, Takakura M,
Nakamura M, Maida Y, Mori N, Bono Y, Sakurai H and Inoue M:
Activation of NF-kappaB is a novel target of KRAS-induced
endometrial carcinogenesis. Clin Cancer Res. 17:1341–1350. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stice JP, Mbai FN, Chen L and Knowlton AA:
Rapid activation of nuclear factor κB by 17β-estradiol and
selective estrogen receptor modulators: Pathways mediating cellular
protection. Shock. 38:128–136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abeyama K, Eng W, Jester JV, Vink AA,
Edelbaum D, Cockerell CJ, Bergstresser PR and Takashima A: A role
for NF-kappaB-dependent gene transactivation in sunburn. J Clin
Invest. 105:1751–1759. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shibata A, Nagaya T, Imai T, Funahashi H,
Nakao A and Seo H: Inhibition of NF-kappaB activity decreases the
VEGF mRNA expression in MDA-MB-231 breast cancer cells. Breast
Cancer Res Treat. 73:237–243. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiong HQ, Abbruzzese JL, Lin E, Wang L,
Zheng L and Xie K: NF-kappaB activity blockade impairs the
angiogenic potential of human pancreatic cancer cells. Int J
Cancer. 108:181–188. 2004. View Article : Google Scholar
|
|
28
|
Stoltz RA, Abraham NG and
Laniado-Schwartzman M: The role of NF-kappaB in the angiogenic
response of coronary microvessel endothelial cells. Proc Natl Acad
Sci USA. 93:2832–2837. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakabayashi H and Shimizu K: Involvement
of Akt/NF-κB pathway in antitumor effects of parthenolide on
glioblastoma cells in vitro and in vivo. BMC Cancer. 12:4532012.
View Article : Google Scholar
|
|
30
|
deGraffenried LA1, Chandrasekar B,
Friedrichs WE, Donzis E, Silva J, Hidalgo M, Freeman JW and Weiss
GR: NF-kappa B inhibition markedly enhances sensitivity of
resistant breast cancer tumor cells to tamoxifen. Ann Oncol.
15:885–890. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stefansson IM, Salvesen HB and Akslen LA:
Vascular proliferation is important for clinical progress of
endometrial cancer. Cancer Res. 66:3303–3309. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dai H, Zhao S, Xu L, Chen A and Dai S:
Expression of Efp, VEGF and bFGF in normal, hyperplastic and
malignant endo-metrial tissue. Oncol Rep. 23:795–799.
2010.PubMed/NCBI
|