Introduction
Gastric cancer is one of the most common epithelial
malignancies and the second leading cause of cancer-related death
worldwide (1). In most patients,
gastric cancer is diagnosed at an advanced stage, accompanied by
extensive invasion and lymphatic metastasis, resulting in the
highest mortality rate among cancers (2). The metastasis of gastric cancer
involves multiple steps and requires the accumulation of the
altered expression of many different genes (3). At present, the mechanisms involved in
gastric cancer metastasis are not fully clear and successful
therapeutic strategies are limited. Therefore, the investigation of
new agents, particularly targeted agents (4), and understanding of the molecular
mechanisms involved in gastric cancer metastasis are vital and may
provide novel avenues for targeted therapy of gastric cancer.
Gastrin, an important gastrointestinal (GI) hormone,
is involved in the stimulation of gastric acid secretion and
epithelial proliferation of the GI tract (5). Most gastrin is synthesized in
antroduodenal G-cells, where progastrin matures to bioactive
carboxyamidated gastrins through multiple modifications (6). The gastrin-17 amide (G-17), accounts
for more than 90% of the G-cell synthesized gastrin in most mammals
(7). In vitro studies have
shown that gastrin stimulates the proliferation of gastric cancer
cell lines through the induction of specific mitogen activated
protein kinase (8). Transgenic mice
overexpressing G-17 were found to exhibit enhanced development and
progression of invasive gastric cancer (9). However, the direct role of gastrin in
the promotion of gastric carcinogenesis remains elusive.
GI peptides, including gastrin and cholecystokinin
(CCK), are members of a structurally diverse group of molecular
messengers that play important roles in the control of appetite and
hormonal secretion (10). The
cholecystokinin-B (CCK-B)/gastrin receptor belongs to the seven
transmembrane G-protein-coupled receptor superfamily. The CCK-B
receptor on the basolateral domain of the cell membrane is
immunoreactive and displays high-affinity binding ability to
gastrin (11). It has been widely
accepted that gastrin, a trophic factor, promotes the growth of
cancer cells both in vitro and in vivo through the
CCK-B receptor, and the expression levels of the gastrin gene and
CCK-B receptor are closely related to the invasiveness of cancer
cells (12). These studies indicate
the need for further research to establish a direct link between
the gastrin/CCK-B receptor pathway and the metastasis of gastric
cancer.
Activation of Wnt signaling involves the inhibition
of β-catenin degradation, resulting in the nuclear accumulation of
β-catenin and transcriptional activation of T-cell
factor/lymphoid-enhancing factor (TCF/LEF) target genes (13). Gastrin has been shown to be a
functionally relevant downstream target of the Wnt signaling
pathway (4). Studies have indicated
that incompletely processed gastrins are capable of inducing
metastatic processes in colon cancers both in vitro and
in vivo (14). Additionally,
Wnt pathway activation was found to contribute to carcinogenesis in
a subset of gastric adenocarcinomas (15). However, the underlying mechanism of
gastrins in regulating gastric cancer metastasis is still
unclear.
In the present study, we aimed to identify the role
of G-17 in the context of gastric cancer. We found that G-17
promoted the expression levels of β-catenin and TCF-4 in gastric
cancer cell lines. In addition, G-17 increased the metastasis of
SGC7901 cells by inducing β-catenin nuclear translocation. Taken
together, our results suggest that G-17 is a pro-metastatic factor,
and may therefore provide a novel therapeutic target for gastric
cancer.
Materials and methods
Cell lines and cultures
Cell plates were pre-covered with Matrigel (BD
Biosciences, Shanghai, China) at 5 µg/cm2. Human
gastric cancer SGC7901 cells were maintained in Dulbecco's modified
Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS)
supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin at
37°C in 5% CO2. SGC7901 cells were divided into four
groups: control group, cells without any treatment; the G-17 group,
cells incubated with 1×10−7 mol/l G-17; the proglumide
(PGL) group, cells incubated with 1×10−7 mol/l PGL,
which is a gastrin receptor antagonist; and the G-17 + PGL group,
cells incubated concurrently with G-17 (1×10−7 mol/l)
and PGL (1×10−7 mol/l).
Western blot analysis
A total of 25 µg of proteins was loaded and
separated via sodium dodecyl sulfate-polyacrylamide gel
electrophoresis, and then electrotransferred to nitrocellulose
membranes (Amersham, Little Chalfont, UK). The membranes were then
blocked in 2.5% non-fat milk for 1 h at 37°C. After washing with
Tris-buffered saline with Tween-20, the membranes were incubated
with primary antibodies against β-catenin, TCF-4, matrix
metalloproteinase (MMP)-7, MMP-9, vascular endothelial growth
factor (VEGF) and β-actin (Santa Cruz Biotechnology, Santa Cruz,
CA, USA) at 4°C overnight. Then, the peroxidase-conjugated
secondary antibody (Boster Corporation, Wuhan, Hubei, China)
diluted in 1:1,000 was added and incubated for 1 h at room
temperature. The immunoreactive protein bands were then visualized
using an enhanced chemiluminescence detection system
(Amersham).
Cell cycle analysis
Cell cycle analysis was determined by flow cytometry
(BD Biosciences, Franklin Lakes, NJ, USA). SGC7901 cells were
harvested, washed with phosphate-buffered saline (PBS), fixed with
75% ethanol overnight at 4°C, and then incubated with RNase at 37°C
for 30 min. Cell nuclei were stained with propidium iodide for 30
min. A total of 104 nuclei were examined in a
FACSCalibur flow cytometer, and DNA histograms were analyzed by
CellQuest software (both from Becton-Dickinson, Mountain View, CA,
USA). Results are presented as the percentage of cells in each
phase.
MTT assay
Cell viability was assessed using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Cells were transfected according to the above description
and were seeded into 96-well plates at 6×103 cells/well.
The surviving fractions were determined at 0, 24, 48, 72, 96 and
120 h. Thereafter, the old medium was discarded and fresh medium
containing MTT (5 mg/ml MTT in PBS; Sangon, Shanghai, China) was
added and incubated for an additional 4 h. Then, cell viability was
measured with a spectrophotometer (Bio-Rad Laboratories, Hercules,
CA, USA) at 470 nm. Each experiment was performed in
triplicate.
Wound healing assay
The CytoSelect 24-well wound healing assay (Cell
Biolabs, Inc., San Diego, CA, USA) was used to analyze the
migration of SGC7901 cells. The assay was performed according to
the manufacturer's recommendations using 2×103
cells/well. Image acquisition of wound fields was carried out after
the removal of inserts (0 h) and wound closure documentation was
completed after 24 h with a phase-contrast microscope (Leica DM IL)
equipped with a digital camera (Leica DFC300FX) (both from Leica
Microsystems, Wetzlar, Germany). Image analysis was conducted by
Adobe Photoshop CS7 software.
Transwell invasion assay
Transwell membranes coated with Matrigel
(Becton-Dickinson, Franklin Lakes, NJ, USA) were used to assay the
invasion of the SGC7901 cells in vitro. Cells were plated at
2×104/well in the upper chamber in serum-free medium,
and 20% FBS was added to the medium in the lower chamber. After
incubation for 24 h, the non-invading cells were removed from the
top well with a cotton swab, while the bottom cells were fixed in
95% ethanol, and stained with hematoxylin. The cell numbers were
determined by counting the penetrating cells under a microscope at
a magnification of ×200 in 10 random fields in each well. Each
experiment was performed in triplicate.
TOP/FOP-FLASH luciferase reporter
assay
The assay was conducted according to a previously
study (16); each well was
incubated with a mixture containing 20 µl of serum-free
DMEM, 0.6 µl of FuGENE, 0.15 µg of the firefly
luciferase reporter plasmid, 0.15 µg of the β-catenin
expression vector, 0.15 µg of the TCF-4 expression vector,
and 0.8 ng of the Renilla luciferase vector phRG-TK. Then,
24 h after transfection, the cells were lysed in 50 µl of
passive lysis buffer, and the luciferase activity was determined
using a luminometer using the Dual Luciferase Assay System
(Promega) on 20 µl of lysate. Results are expressed as fold
induction. Fold induction was determined by normalizing each
firefly luciferase value to the Renilla luciferase internal
control value and by dividing these normalized values with the mean
normalized value of the corresponding reporter construct
transfected with the empty expression vectors.
Immunofluorescence staining
Fluorescent cells were cultured on a 8-well chamber
CultureSlides (Becton-Dickinson, Bedford, MA, USA). After 8 h, the
cells were fixed in 3% paraformaldehyde in PBS at room temperature
for 8 min, then permeabilized with 0.2% Triton X-100 for 15 min at
room temperature. After washing in PBS, the cells were incubated
with primary mouse anti-β-catenin monoclonal antibody (1 mg/ml;
Transduction Laboratories, Lexington, KY, USA) at 4°C overnight.
After washing, the cells were incubated with biotinylated goat
anti-mouse IgG (Pierce, Rockford, IL, USA) at room temperature for
1 h. The immunoreactivity was revealed using Alexa 568-conjugated
streptavidin (Molecular Probes, Eugene, OR, USA), and the cells
were counterstained with 10 mg/ml 4′,6-diamidino-2-phenylindole
dihydrochloride (DAPI). The cells were examined under a Nikon
fluorescence microscope (Image Systems, Columbia, MD, USA).
Statistical analysis
All results are presented as mean ± SD. The
statistical significance of the studies was analyzed using the
Student's t-test. The difference was considered statistically
significant at P<0.05.
Results
G-17 accelerates cell cycle progression
and proliferation of SGC7901 cells
To investigate the impact of G-17 on the SGC7901
cells, we studied whether G-17 was capable of affecting the cell
cycle and proliferation. Flow cytometric analysis indicated that
the G0/G1 phase in the SGC7901 cells was accelerated in the G-17
group, and an accumulation of G0/G1 phase in the PGL group compared
with the control group. However, the G0/G1 phase was shortened in
the G-17 + PGL group compared with the PGL group (Fig. 1A and B). These results indicated an
activation of G0/G1 progression in the SGC7901 cells following
incubation with G-17. The cell proliferation assay was performed in
SGC7901 cells; G-17 was observed to strongly increase the cell
growth compared with the control group, and the decreased cell
numbers caused by PGL were balanced by G-17 treatment (P<0.05)
(Fig. 1C). These findings
demonstrated that G-17 promoted the cell cycle by accelerating the
G0/G1 phase and increasing cell proliferation levels in the SGC7901
cells.
G-17 increases the migration and invasion
of SGC7901 cells
Given the impact of G-17 on the cell cycle and
proliferation of SGC7901 cells, scratch assays were next carried
out to measure the motility of the SGC7901 cells induced by G-17.
The G-17 group showed an almost complete closure of the gap,
whereas the PGL group reduced the gap by only ~20% compared with
that noted in the control group (P<0.05), indicating that G-17
promoted the migration of SGC7901 cells via binding to its
receptor. In addition, the wound closed by nearly 75% in the G-17 +
PGL group (P<0.05) (Fig. 2A and
B), indicating the antagonistic effect of G-17 towards PGL. The
results of the Transwell assay were coincident with the results of
the scratch assays. The number of invasive SGC7901 cells was
increased by G-17, but decreased by PGL compared with the control
group, and the motile ability was recovered in the G-17 + PGL group
compared with that noted in the PGL group (P<0.05) (Fig. 2C and D). These observations suggest
that G-17 is a positive metastatic regulator of gastric cancer.
G-17 promotes the expression levels of
cancer metastasis-associated proteins
The expression levels of MMP-7, MMP-9 and VEGF were
determined by western blotting. The results indicated that the
levels of the three proteins were increased in the G-17 group and
decreased in the PGL group compared with these levels in the
control group (P<0.05), while the G-17 + PGL group exhibited
higher expression levels of MMP-7, MMP-9 and VEGF in comparison
with the PGL group (P<0.05) (Fig.
3). The increased expression of migration-related proteins
further confirmed of the effect of G-17, which promotes the
metastasis of SGC7901 cells.
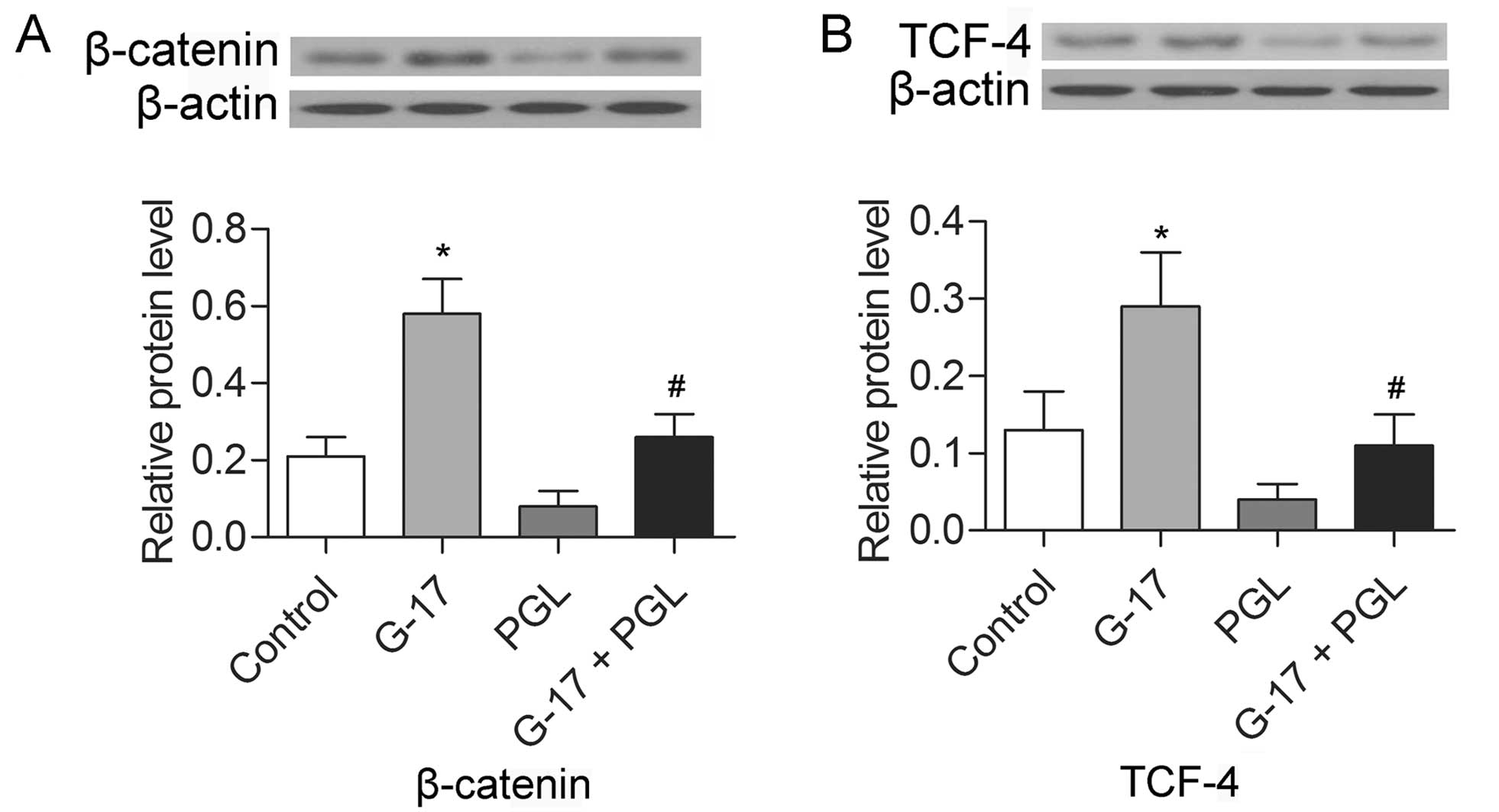
G-17 promotes the expression of β-catenin
and TCF-4
The expression levels of β-catenin and TCF-4 were
detected in the SGC7901 cells by western blotting. The results
indicated that the levels of both β-catenin and TCF-4 were
increased in the G-17 group and decreased in the PGL group compared
with the levels in the control group (P<0.05), while the G-17 +
PGL group exhibited higher expression levels of β-catenin and TCF-4
in comparison with levels noted in the PGL group (P<0.05)
(Fig. 4). The results indicate that
G-17 induced the activation of the β-catenin pathway via binding to
the G-17 receptor in the SGC7901 cells.
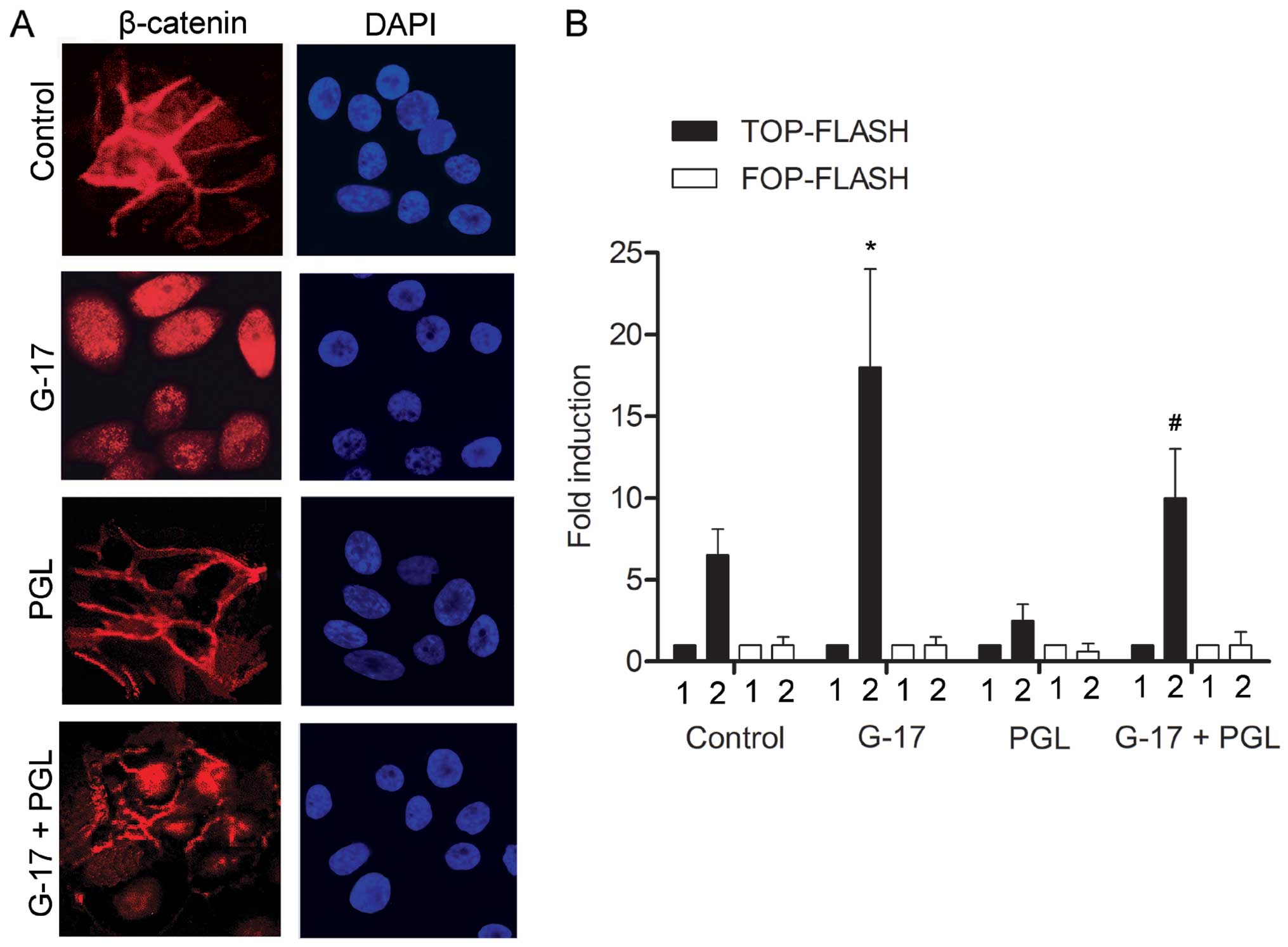
G-17 induces the β-catenin nuclear
translocation in SGC7901 cells
To further explore the mechanism involved in the
metastasis of SGC7901 cells by G-17, the subcellular localization
of β-catenin was evaluated. The G-17 group displayed predominantly
nuclear β-catenin staining. Cells in the PGL group showed
membranous β-catenin staining with minimal cytoplasmic or nuclear
staining, whereas cells in the G-17 + PGL group displayed more
cytoplasmic and nuclear staining of β-catenin as compared with that
noted in PGL group (Fig. 5A). We
then compared the ability of SGC7901 cells in each group to
transactivate the luciferase reporter plasmid containing the
wild-type (TOP-FLASH) or mutated (FOP-FLASH) β-catenin/TCF binding
site as regulatory elements. Cells induced by G-17 displayed a
higher ability to transactivation the TOP-FLASH reporter plasmid
than the control group (Fig. 5B).
The G-17 + PGL group exhibited a higher TOP-FLASH transactivate
activity compared with the PGL group. In accordance with our
findings, showing a preferential cytoplasmic and nuclear
localization of β-catenin in the G-17-induced cells, these data
clearly indicate that G-17-treated cells had stronger β-catenin/TCF
transcriptional activity than the untreated cells, and that G-17
promoted the nuclear translocation of β-catenin to activate the Wnt
signaling pathway in the SGC7901 cells.
The β-catenin/TCF-4 pathway mediates
G-17-induced metastasis of SGC7901 cells
The former experiment provided convincing evidence
that G-17 promotes metastasis by activating the β-catenin/TCF-4
pathway; however, it is unclear whether the β-catenin/TCF-4 pathway
is necessary for the G-17-induced metastasis of SGC7901 cells. Axin
is known to inhibit the Wnt-signaling pathway via facilitating the
phosphorylation and thus the degradation of β-catenin (17). The results showed that the migratory
and invasive abilities of the SGC7901 cells were suppressed by axin
compared with these parameters in the control group (P<0.05)
(Fig. 6A–D), suggesting the
indispensable role of β-catenin in G-17-induced metastasis of
SGC7901 cells. The motile ability was decreased in the G-17 + axin
group compared with that noted in the G-17 group (P<0.05),
indicating that the G-17-induced metastasis in SGC7901 cells relies
on β-catenin. The expression levels of β-catenin, TCF-4, MMP-7,
MMP-9 and VEGF were strongly inhibited by axin compared with the
control group. The inhibitory effect in the G-17 + axin group was
detected compared with the G-17 group (Fig. 6E–G), further showing that the
G-17-induced metastasis was dependent on the β-catenin/TCF-4
pathway in the SGC7901 cells. These results indicated that the
β-catenin/TCF-4 pathway is essential for mediating G-17-induced
metastasis in SGC7901 cells.
 | Figure 6The β-catenin/TCF-4 pathway mediates
the G-17-induced metastasis of SGC7901 cells. SGC7901 cells were
divided into four groups: the control group, cells without any
treatment; the G-17 group, cells incubated with 1×10−7
mol/l G-17; axin group, cells incubated with 1 µg axin; and
the G-17 + PGL group, cells concurrently incubated with G-17
(1×10−7 mol/l) and 1 µg axin. (A) Wound-healing
assays of SGC7901 cells treated with G-17 or/and axin for 24 h. (B)
Histogram showing the quantification of wound-healing assays of
SGC7901 cells in each group. (C) Transwell assays of SGC7901 cells
treated with G-17 or/and axin for 48 h. (D) Histogram illustrating
the quantification of Transwell assays of SGC7901 cells in each
group. (E) The expression levels of β-catenin, TCF-4, MMP-7, MMP-9
and VEGF were detected in the SGC7901 cells by western blotting.
Relative protein expression of (F) β-catenin, TCF-4, (G) MMP-7,
MMP-9 and VEGF were quantified using Image-Pro Plus 6.0 software
and normalized to β-actin. Data are represented as the mean ± SD of
three experiments; *P<0.05 vs. the control group,
#P<0.05 vs. the G-17 group. |
Discussion
Gastric cancer is the second leading cause of
cancer-related deaths worldwide (18). Gastric cancer metastasis is a
crucial factor in the determination of the clinical staging,
prognosis and survival of gastric cancer patients (19). Therefore, identifying metastatic
factors and elucidating the molecular mechanisms underlying gastric
cancer metastasis have become critical issues. Accumulated evidence
indicates that G-17 has growth-promoting and oncogenic function in
gastric cancers (20,21); its role in metastasis and the
relative mechanism in gastric cancer were explored in the present
study.
The CCK-B receptor is widely distributed throughout
the human GI tract and mediates the normal physiological function
of gastrin. Gastrin has proliferative effects on various
malignancies including gastric and colorectal cancers through the
CCK-B receptor (22,23). Gastrin and CCK have been reported to
mediate the proliferative responses in a variety of normal and
cancer cell model systems (20).
Gastrin in particular has been implicated in accelerating the
development of gastrointestinal cancers (24). Previously, Pradeep et al
reported that G-17 induced the activation of G1 progression in
gastric adenocarcinoma cell line AGSE (25). To investigate the G-17 function in
gastric cancer via binding to its receptor, a gastrin receptor
antagonist, PGL was used in this research. The results indicated an
activation of G1 progression following incubation with G-17, but a
retardation in the G0/G1 phase with PGL. Our results demonstrated
that G-17 promoted cell cycle progression by accelerating the G0/G1
phase via binding to the gastrin receptor in SGC7901 cells.
Metastasis is an important element of gastric cancer
progression, and leads to a high mortality rate and poor prognosis
(26). Cell invasive ability is
critical for tumor metastasis (27). In the present study, the migratory
and invasive abilities of the SGC7901 cells were increased
following treatment with G-17, whereas they were decreased by PGL,
suggesting that G-17 is a metastasis-associated biomarker in
gastric cancer. Apart from G-17, microRNA-335 (28) and SOX-2 (29) were reported to be suppressors, while
Y-box binding protein-1 was indicated as a promoter in the
metastasis of gastric cancer (30).
These findings, along with ours, indicate that potential prognostic
biomarkers could serve as potential gene targets in gastric cancer
metastasis treatment.
MMPs have been recognized as the most important
protease family, and mediate central events in tumor progression,
including invasion, metastasis and angiogenesis (31). Higher MMP-7 and MMP-9 levels were
reported to be associated with the invasive phenotype of gastric
cancer (32,33). VEGF is a well-defined pro-angiogenic
and pro-metastatic factor in numerous human cancers (34), and the upregulation of VEGF
contributes to the tumor invasion and metastasis of gastric cancer
(35). The expression levels of
MMP-7, MMP-9 and VEGF were significantly increased by G-17, but
were inhibited by PGL in the present study. The results provide
further evidence of the metastasis-promoting role of G-17 in
gastric cancer via binding to the gastrin receptor.
Studies indicate that β-catenin and TCF4, which are
important genes in the Wnt pathway, are critical oncogenes in
GI-related tumorigenesis (36).
Thus, we evaluated the expression of β-catenin and TCF-4 in the
SGC7901 cells. The results revealed that G-17 induced the
activation of the β-catenin pathway via binding to the gastrin
receptor in the SGC7901 cells. Higher β-catenin/TCF activity
induced by G-17 was measured by the TOP-/FOP-FLASH luciferase
reporter assay, strongly supporting a role of G-17 in activating
the β-catenin/TCF pathway in gastric cancer. Nuclear β-catenin has
been reported to be the hallmark of an active Wnt pathway (37). The stimulation of β-catenin nuclear
translocation further confirmed the suppressive role of G-17 in
β-catenin degradation. These results were coincident with the
behavior of gastrin in colorectal tumor cells, in which it
prolonged the t1/2 of β-catenin protein by stabilizing
β-catenin (38), and demonstrated
that gastrin appears to exert the nuclear translocation of
β-catenin to activate the Wnt pathway in gastric cancer.
Axin is known to inhibit the Wnt-signaling pathway
via facilitating the phosphorylation and the degradation of
β-catenin. To investigate the necessity of the Wnt pathway in
G-17-induced gastric cancer metastasis, G-17 was incubated with or
without axin in the present study. The results showed that the
migratory and invasive abilities of the SGC7901 cells were
suppressed by axin, but recovered to some extent by G-17. These
results indicated that the β-catenin/TCF-4 pathway is essential in
mediating G-17-induced metastasis in gastric cancer, and G-17
exhibited antagonism to the β-catenin degradation by axin. The
present study indicated that the APC/axin/GSK complex is a
degradation agent of β-catenin; the dissociation of the complex
leads to β-catenin accumulation. The free-β-catenin then
translocates to the nucleus where it binds to T-cell factors and
activates the transcription of a number of genes, including c-Myc,
cyclin D1 and MMP-7 (37). Compared
with the effect of axin in the present study, the expression levels
of β-catenin, TCF-4, MMP-7, MMP-9 and VEGF in the axin + G-17 group
were enhanced, and the results confirmed the suppressive role of
G-17 in β-catenin degradation in gastric cancer.
Along with these data, our present research explored
an important GI hormone, G-17, in gastric cancer SGC7901 cells.
Through binding to the gastrin receptor, G-17 promoted cell
progression and metastasis, and activated the β-catenin/TCF-4
pathway. In addition, the β-catenin/TCF-4 pathway was found to be
essential for mediating G-17-induced metastasis and G-17 suppressed
the β-catenin degradation in gastric cancer. These results offer a
potential gene target for the treatment of gastric cancer.
Abbreviations:
|
G-17
|
gastrin-17 amide
|
|
CCK-B
|
cholecystokinin-B
|
|
PGL
|
proglumide
|
|
TCF
|
T-cell factor
|
|
MMP
|
matrix metalloproteinase
|
|
VEGF
|
vascular endothelial growth factor
|
Acknowledgments
The authors would like to thank the members of the
Second Affiliated Hospital of Xi'an Jiaotong University and Xi'an
Central Hospital, for providing technical support and helpful
discussions concerning the present study.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Coburn NG: Lymph nodes and gastric cancer.
J Surg Oncol. 99:199–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hippo Y, Yashiro M, Ishii M, Taniguchi H,
Tsutsumi S, Hirakawa K, Kodama T and Aburatani H: Differential gene
expression profiles of scirrhous gastric cancer cells with high
metastatic potential to peritoneum or lymph nodes. Cancer Res.
61:889–895. 2001.PubMed/NCBI
|
|
4
|
Doi T, Muro K, Boku N, Yamada Y, Nishina
T, Takiuchi H, Komatsu Y, Hamamoto Y, Ohno N, Fujita Y, et al:
Multicenter phase II study of everolimus in patients with
previously treated metastatic gastric cancer. J Clin Oncol.
28:1904–1910. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Friis-Hansen L, Sundler F, Li Y, Gillespie
PJ, Saunders TL, Greenson JK, Owyang C, Rehfeld JF and Samuelson
LC: Impaired gastric acid secretion in gastrin-deficient mice. Am J
Physiol. 274:G561–G568. 1998.PubMed/NCBI
|
|
6
|
Rehfeld JF, Lindberg I and Friis-Hansen L:
Progastrin processing differs in 7B2 and PC2 knockout animals: A
role for 7B2 independent of action on PC2. FEBS Lett. 510:89–93.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rehfeld JF, Hansen CP and Johnsen AH:
Post-poly(Glu) cleavage and degradation modified by O-sulfated
tyrosine: A novel post-translational processing mechanism. EMBO J.
14:389–396. 1995.PubMed/NCBI
|
|
8
|
Gutkind JS: The pathways connecting G
protein-coupled receptors to the nucleus through divergent
mitogen-activated protein kinase cascades. J Biol Chem.
273:1839–1842. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang TC, Dangler CA, Chen D, Goldenring
JR, Koh T, Raychowdhury R, Coffey RJ, Ito S, Varro A, Dockray GJ,
et al: Synergistic interaction between hypergastrinemia and
Helicobacter infection in a mouse model of gastric cancer.
Gastroenterology. 118:36–47. 2000. View Article : Google Scholar
|
|
10
|
Rozengurt E and Walsh JH: Gastrin, CCK,
signaling, and cancer. Annu Rev Physiol. 63:49–76. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kulaksiz H, Arnold R, Göke B, Maronde E,
Meyer M, Fahrenholz F, Forssmann WG and Eissele R: Expression and
cell-specific localization of the cholecystokinin B/gastrin
receptor in the human stomach. Cell Tissue Res. 299:289–298. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Watson SA, Morris TM, McWilliams DF,
Harris J, Evans S, Smith A and Clarke PA: Potential role of
endocrine gastrin in the colonic adenoma carcinoma sequence. Br J
Cancer. 87:567–573. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park WS, Oh RR, Park JY, Lee SH, Shin MS,
Kim YS, Kim SY, Lee HK, Kim PJ, Oh ST, et al: Frequent somatic
mutations of the β-catenin gene in intestinal-type gastric cancer.
Cancer Res. 59:4257–4260. 1999.PubMed/NCBI
|
|
14
|
Kermorgant S and Lehy T: Glycine-extended
gastrin promotes the invasiveness of human colon cancer cells.
Biochem Biophys Res Commun. 285:136–141. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Clements WM, Wang J, Sarnaik A, Kim OJ,
MacDonald J, Fenoglio-Preiser C, Groden J and Lowy AM: β-Catenin
mutation is a frequent cause of Wnt pathway activation in gastric
cancer. Cancer Res. 62:3503–3506. 2002.PubMed/NCBI
|
|
16
|
Gilles C, Polette M, Mestdagt M,
Nawrocki-Raby B, Ruggeri P, Birembaut P and Foidart JM:
Transactivation of vimentin by β-catenin in human breast cancer
cells. Cancer Res. 63:2658–2664. 2003.PubMed/NCBI
|
|
17
|
Kikuchi A: Modulation of Wnt signaling by
Axin and Axil. Cytokine Growth Factor Rev. 10:255–265. 1999.
View Article : Google Scholar
|
|
18
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang ZG, Gao L, Guo XB and Shi YL: Roles
of long non-coding RNAs in gastric cancer metastasis. World J
Gastroenterol. 21:5220–5230. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Basu A, Sondarva G, Santha S, Rana A and
Rana B: Elucidation of signaling pathways that mediate
gastrin-induced JNK activation and pGSK3β/Snail induction in
gastric cancer cells. Cancer Res. 75(Suppl 15): 19672015.
View Article : Google Scholar
|
|
21
|
Yakut M, Örmeci N, Erdal H, Keskin O,
Karayel Z, Tutkak H and Soykan I: The association between
precancerous gastric lesions and serum pepsinogens, serum gastrin,
vascular endothelial growth factor, serum interleukin-1 Beta, serum
toll-like receptor-4 levels and Helicobacter pylori Cag A status.
Clin Res Hepatol Gastroenterol. 37:302–311. 2013. View Article : Google Scholar
|
|
22
|
Han YM, Park JM, Park SH, Hahm KB, Hong SP
and Kim EH: Gastrin promotes intestinal polyposis through
cholecystokinin-B receptor-mediated proliferative signaling and
fostering tumor microenvironment. J Physiol Pharmacol. 64:429–437.
2013.PubMed/NCBI
|
|
23
|
Huang BP, Lin CH, Chen YC and Kao SH:
Expression of cholecystokinin receptors in colon cancer and the
clinical correlation in Taiwan. Tumour Biol. 37:4579–4584. 2016.
View Article : Google Scholar
|
|
24
|
Marshall KM, Laval M, Estacio O, Hudson
DF, Kalitsis P, Shulkes A, Baldwin GS and Patel O: Activation by
zinc of the human gastrin gene promoter in colon cancer cells in
vitro and in vivo. Metallomics. 7:1390–1398. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pradeep A, Sharma C, Sathyanarayana P,
Albanese C, Fleming JV, Wang TC, Wolfe MM, Baker KM, Pestell RG and
Rana B: Gastrin-mediated activation of cyclin D1 transcription
involves β-catenin and CREB pathways in gastric cancer cells.
Oncogene. 23:3689–3699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jie D, Zhongmin Z, Guoqing L, Sheng L, Yi
Z, Jing W and Liang Z: Positive expression of LSD1 and negative
expression of E-cadherin correlate with metastasis and poor
prognosis of colon cancer. Dig Dis Sci. 58:1581–1589. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pang MF and Nelson CM: Intercellular
communication, the tumor microenvironment, and tumor progression.
Intercellular Communication in Cancer. Springer; pp. 343–362. 2015,
View Article : Google Scholar
|
|
28
|
Xu Y, Zhao F, Wang Z, Song Y, Luo Y, Zhang
X, Jiang L, Sun Z, Miao Z and Xu H: MicroRNA-335 acts as a
metastasis suppressor in gastric cancer by targeting Bcl-w and
specificity protein 1. Oncogene. 31:1398–1407. 2012. View Article : Google Scholar :
|
|
29
|
Wang S, Tie J, Wang R, Hu F, Gao L, Wang
W, Wang L, Li Z, Hu S, Tang S, et al: SOX2, a predictor of survival
in gastric cancer, inhibits cell proliferation and metastasis by
regulating PTEN. Cancer Lett. 358:210–219. 2015. View Article : Google Scholar
|
|
30
|
Guo T, Yu Y, Yip GWC, Baeg GH, Thike AA,
Lim TK, Tan PH, Matsumoto K and Bay BH: Y-box binding protein 1 is
correlated with lymph node metastasis in intestinal-type gastric
cancer. Histopathology. 66:491–499. 2015. View Article : Google Scholar
|
|
31
|
Egeblad M and Werb Z: New functions for
the matrix metalloproteinases in cancer progression. Nat Rev
Cancer. 2:161–174. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Z, Zhang D, Zhang H, Miao Z, Tang Y,
Sun G and Dai D: Prediction of peritoneal recurrence by the mRNA
level of CEA and MMP-7 in peritoneal lavage of gastric cancer
patients. Tumour Biol. 35:3463–3470. 2014. View Article : Google Scholar
|
|
33
|
Shimjura T, Dagher A, Ebi M, Yamada T,
Yamada T, Joh T and Moses MA: Potential of urinary MMP-9/NGAL
complex as a novel biomarker for the early detection of gastric
cancer. Cancer Res. 74(Suppl 19): S892. 2014. View Article : Google Scholar
|
|
34
|
Tsai CY, Wang CS, Tsai MM, Chi HC, Cheng
WL, Tseng YH, Chen CY, Lin CD, Wu JI, Wang LH, et al:
Interleukin-32 increases human gastric cancer cell invasion
associated with tumor progression and metastasis. Clin Cancer Res.
20:2276–2288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Koh SA, Kim MK, Lee KH, Kim SW and Kim
J-R: RhoGDI2 is associated with HGF-mediated tumor invasion through
VEGF in stomach cancer. Clin Exp Metastasis. 31:805–815. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bienz M and Clevers H: Linking colorectal
cancer to Wnt signaling. Cell. 103:311–320. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Doucas H, Garcea G, Neal CP, Manson MM and
Berry DP: Changes in the Wnt signalling pathway in gastrointestinal
cancers and their prognostic significance. Eur J Cancer.
41:365–379. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Song DH, Kaufman JC, Borodyansky L,
Albanese C, Pestell RG and Wolfe MM: Gastrin stabilises β-catenin
protein in mouse colorectal cancer cells. Br J Cancer.
92:1581–1587. 2005. View Article : Google Scholar : PubMed/NCBI
|