Introduction
Since microRNAs (miRs) were first discovered in the
early 1990s, their functional roles in regulating cellular
physiology of both normal and cancerous cells have been highlighted
by many studies (1). In cancer
cells, miR dysregulation was first identified in a study on chronic
lymphocytic leukemia (CLL), in which mir-15 and mir-16 were
frequently deleted at the chromosome locus 13q14.3 (2). This deletion induced upregulation of
BCL-2, which led to anti-apoptotic advantages to the cells. It has
been accepted that miRs can serve as either oncogenes or tumor
suppressor genes. During transformation of normal cells, oncogenic
miRs are often overexpressed, while tumor-suppressing miRs are
downregulated (3–5). The expression profile of miRs in each
tumor type is unique, and many researchers have therefore attempted
to evaluate their clinical significance as diagnostic tools
(6–8). For example, Puerta-Gil and colleagues
reported that miRs in urine, such as mir-143, mir-222, and mir-452,
might be useful diagnostic markers for bladder cancer (9). Wang et al also demonstrated
that mir-141 is associated with diagnosis of bladder cancer using
114 bladder tumors and matched normal bladder tissues (10). Recently, clinical trials using miRs
as therapeutics have gained popularity; 138 clinical trials related
to miRs are ongoing or have been completed to date (https://clinical-trials.gov/). Although bladder cancer
is one of the most fatal diseases in Western countries, sufficient
clinical studies on bladder cancer have not yet been performed.
This might be due in part to the heterogeneity of the disease and
the absence of signature mutations (11). Therefore, in bladder cancers,
identifying effective molecular targets is critical. Iyer and
coworkers attempted to verify genetic alterations in bladder
cancers using 97 advanced-stage bladder tumors. In their
integrative analysis, driver mutations in the RTK-RAS-RAF axis, the
PI3K/AKT/mTOR axis, and in regulators of G1/S cell cycle phases
were identified (12). In
accordance, our previous study verified that mir-20b regulates the
growth and migration of bladder cancer cells through cell cycle
accumulation in G1/S phase and matrix metalloproteinase (MMP)
regulation (13).
In this study, we report the novel finding that
mir-106a regulates the proliferation and migration of bladder
cancer cells via modulating MAPKs (ERK p38, and JNK),
p21CIP1/WAF1/cyclin D1/CDK 6, and Ets-1/MMP-2.
Materials and methods
Materials
Antibodies for ERK, phospho-ERK, p38, phospho-p38,
JNK, and phospho-JNK were purchased from Cell Signaling Technology
(Danvers, MA, USA). Polyclonal antibodies against cyclin D1, cyclin
E, CDK2, CDK6, p53, p21CIP1/WAF1, p27KIP1,
GAPDH, Ets-1, Sp-1, ATF-2, and CREB were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). The anti-MMP-2 antibody was
obtained from Chemicon International (Billerica, MA, USA). mir-106a
(5′-AAAAGuGCuuACAGUGCAGGuAG-3′) and mir-106a inhibitor were
obtained from Genolution (Seoul, Korea).
Cell cultures
Human bladder carcinoma cell lines (EJ, 5637, and
T24) were purchased from the American Type Culture Collection
(ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% fetal calf
serum, l-glutamine, and antibiotics (Biological Industries, Beit
Haemek, Israel) at 37°C in a 5% CO2 humidified
incubator. Normal human urothelial cells (HUCs) were purchased from
ScienCell Research Laboratories (Carlsbad, CA, USA). The cells were
grown in the medium specific for HUCs with supplements according to
the manufacturer's protocol.
Quantitative real-time RT-PCR
(qRT-PCR)
MicroRNA expression was measured using a Rotor-Gene
6000. Real-time PCR assays were performed using a miScript PCR
Starter kit (Qiagen Korea, Seoul, Korea) as previously described
(14). RT-PCR conditions were as
follows: 1X initial enzyme activation for 15 min at 95°C, then 50
cycles of denaturation for 15 sec (94°C), annealing for 30 sec
(55°C) and extension for 30 sec (70°C). The melt curve was
performed from 70–99°C at a heating rate of 1°C/5 sec. Spectral
data were analyzed using Rotor-Gene Real-Time Analysis Software 6.0
Build 14. All experiments were performed in triplicate. Expression
of miRNAs was normalized to U6 RNA.
Bioinformatics analysis
Screening of putative targets of mir-106a was
performed by utilizing the miRanda algorithm (http://www.microrna.org/microrna/home.do) and the NCBI
mRNA database (NCBI mRNA DB: http://www.ncbi.nlm.nih.gov/).
Cell proliferation
Cell proliferation was studied using
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT)
assay as previously described (15). Cellular morphology images were
obtained using phase-contrast microscopy.
Transfection
Cells were transfected with mir-106a and its
inhibitor using Lipofectamine 2000 transfection reagent (Invitrogen
Corp., Carlsbad, CA, USA) according to the manufacturer's protocol.
After transfection for 48 h, cells were subjected to further
experiments including MTT, immunoblotting, invasion, wound-healing
migration, zymography, and electrophoretic mobility shift assays
(EMSA).
Flow cytometry cell cycle analysis
Cells were harvested and fixed in 70% ethanol. After
washing the cells with 1X ice-cold PBS, they were treated with
RNase (1 mg/ml) followed by propidium iodide (50 mg/ml). Cell cycle
phase distribution was analyzed using a Becton-Dickinson FACStar
flow cytometer equipped with Becton-Dickinson Cell Fit
software.
Immunoblot analysis
Preparation of cell lysates and measurement of
protein concentrations were performed as described previously
(13). Lysates were then
electrophoresed on 10% SDS polyacrylamide gels (SDS-PAGE) under
denaturing conditions and transferred to nitrocellulose membranes
(Hybond, Amersham Corp.). Membranes were blocked with 5% (w/v)
non-fat dry milk in TBS [10 mM Tris-HCl (pH 8.0), 150 mM NaCl]
followed by incubation with primary antibodies at 4°C overnight.
Then, blots were incubated with secondary antibodies for 90 min.
Detection was performed using a Chemiluminescence reagent kit
(Amersham Corp.). Experiments were repeated at least thrice.
Immunoprecipitation and immune complex
kinase assays
Cell lysates were collected from cell pellets using
ice-cold lysis buffer. Briefly, cell lysates were centrifuged at
10,000 × g for 5 min. The supernatants were precipitated by
protein-A sepharose beads pre-coated with indicated antibodies at
4°C for 2 h. Then the beads were washed 4 times with 1 ml lysis
buffer and twice with a kinase buffer as previously described
(13). Finally, pellets were
resuspended in 25 µl of the kinase buffer containing 1
µg of glutathione S-transferase (GST)-pRb C-terminal (pRb
amino acids 769-921) fusion protein (Santa Cruz Biotechnology), 20
µM ATP, and 5 µCi of [γ32P]-ATP (4,500
µCi/mmol; ICN). Subsequently, resuspended pellets were
incubated for 20 min at 30°C with occasional mixing. The kinase
reactions were terminated by the addition of 25 µl of 2X
Laemmli sample buffer and were heated at 100°C for 5 min. Samples
were resolved on 10% SDS-polyacrylamide gels, and then the gels
were dried. Radioactive bands were detected on a film.
Wound-healing migration assay
Cells (3×105) were seeded per well in
6-well plates. Monolayers were scratched with a 2-mm-wide pipette
tip. After washing three times with 1X PBS, plates were incubated
at 37°C in serum-free medium. Migration of cells into the scratched
area was visualized under an inverted microscope at ×40
magnification.
Invasion assay
Invasion assays were performed using an invasion
assay kit (Cell Biolabs, USA) according to the manufacturer's
instructions. Cells (2.5×104) were resuspended in
serum-free medium and plated in the upper chamber of the apparatus.
Medium with 10% FBS was added to the lower chamber as a
chemoattractant. After incubation for 24 h, cells in the lower
chamber were fixed, stained, and photographed. The invasion
potential of the cells was estimated using a commercial cell
invasion assay kit (Chemicon International).
Gelatin zymography
Conditioned cell culture media was electrophoresed
in a polyacrylamide gel containing 1 mg/ml gelatin. The gel was
then washed with 2.5% Triton X-100 for 2 h at room temperature,
followed by overnight incubation at 37°C with a buffer containing
10 mM CaCl2, 150 mM NaCl and 50 mM Tris-HCl (pH 7.5).
The gel was photographed on a light box after staining with 0.2%
Coomassie blue. Areas of gelatinase activity were verified as a
clear band in a dark blue field.
Preparation of nuclear extracts and
EMSA
Nuclear proteins were prepared as described
previously (16). Briefly, cells
were washed, scraped, and resuspended in a buffer containing 10 mM
HEPES (pH 7.9), 10 mM KCl, 1 mM DTT, 0.5 mM PMSF, 0.1 mM EDTA, and
0.1 mM EGTA. Then the cells were lysed, homogenized, and
centrifuged. The nuclear pellets were extracted with an ice-cold
high salt buffer containing 0.4 M NaCl, 20 mM HEPES (pH 7.9), 1 mM
EDTA, 1 mM EGTA, 1 mM DTT, and 1 mM PMSF. After centrifugation,
supernatants were collected. Protein concentration was measured
using the Bradford reagent method (Bio-Rad). Electrophoretic
mobility shift assay (EMSA) was accomplished as previously
described (16). Briefly, the
oligonucleotides spanning the MMP-2 cis element of interest
were end-labeled with 32P-ATP by T4 polynucleotide
kinase (Promega, Madison, WI, USA). The nuclear extracts were
incubated with a radiolabeled oligonucleotide probe (10,000 cpm)
for 20 min at 4°C in a binding buffer containing 25 mM HEPES (pH
7.9), 50 mM NaCl, 0.5 mM EDTA, 0.5 mM DTT, 2.5% glycerol, and 2
µg of poly dI/dC. The DNA-protein complexes were separated
on 6% SDS-PAGE gels at 4°C with TBE running buffer (89 mM Tris, 89
mM boric acid, and 1 mM EDTA). The gel was washed, dried, and then
exposed to X-ray film for 10 h. The sequences for the
oligonucleotides were as follows: Ets-1, GATCTCGAGCCGGAAGTTCGA;
Sp-1, GCCCATTCCTTCCGCCCCC AGATGAAGCAG; ATF-2, AGAGATTGCCTGA CGTCAGA
GAGCTAG; CREB, AGAGATTGCCTGACGTCA GAGAGC TAG; and C/EBP,
TGCAGATTGCGCAATCTGCA. For the competition of Ets-1, Sp-1, ATF-2,
and CREB, each specific polyclonal antibody was supplied to the
binding reaction prior to the incubation of radiolabeled probe.
Statistical analysis
Where appropriate, data are represented as the means
± SE. Data were evaluated by factorial ANOVA and a Fisher's least
significant difference test where appropriate. Statistical
significance was considered at P<0.05.
Results
Expression of mir-106a is downregulated
in bladder cancer cells
In order to investigate the role of mir-106a in
bladder cancers, we first measured the basal expression levels of
mir-106a in bladder cancer cell lines EJ, T-24, and 5637, and
compared their miR expression with normal human urothelial cells
(HUC). Quantitative real-time PCR results showed that all bladder
cancer lines tested exhibited significantly lower levels of
mir-106a than HUC cells (Fig. 1A).
Because mir-106a expression was minimal in EJ cells, ~20% of the
expression seen in HUCs, EJ cells were subjected to further
experiments. We next assessed the effect of mir-106a on the
proliferation of EJ bladder cancer cells by transfecting a mir-106a
mimic, a mir-106a inhibitor, or Lipofectamine 2000 only (control).
As seen in Fig. 1B, transfection of
mir-106a significantly inhibited the proliferation of EJ cells in a
dose-dependent manner. Control or inhibitor treatment elicited no
changes in cell growth. In accordance, mir-106a-transfected EJ
cells showed a dose-dependent decrease in cell viability (Fig. 1C). Based on the cell growth study,
subsequent experiments were performed using 30 nM (around the
IC50) of mir-106a. We then hypothesized that
transfection of miR106a into EJ cancer cells may modulate molecular
effectors associated with proliferation. To narrow the candidates,
we utilized the microRNA bioinformatics database miRanda
(http://www.microrna.org/microrna.home.do). Nucleotide
sequence alignment suggested that mitogen-activated protein kinases
(MAPKs) including ERK, p38, and JNK might be targets of mir-106a
(Fig. 1E). To validate this
screening result, we performed immunoblot assays to determine
whether the MAPKs were modulated by the transfection of mir-106a.
Interestingly, introduction of mir-106a into EJ cancer cells led to
a downregulation of ERK phosphorylation, while p38 phosphorylation
was increased >2-fold and JNK phosphorylation was increased
moderately (Fig. 1D).
mir-106a leads to G1-phase cell cycle
arrest in bladder cancer cells
We speculated that the growth inhibition of bladder
cancer cells induced by mir-106a might be due to cell cycle arrest.
Therefore, we investigated cell cycle phase distribution in
mir-106a-transfected cells using flow cytometry. As demonstrated in
the DNA histograms, mir-106a resulted in an accumulation in the G1
phase of the cell cycle. However, control cells and cells treated
with the mir-106a inhibitor showed no changes in cell cycle
distribution (Fig. 2A–C). In
accordance with the accumulation of cells in G1, the number of
cells in S-phase was reduced (Fig.
2D).
mir-106a targets cyclin D1/CDK6 leading
to upregulation of p21CIP1/WAF1in bladder cancer
cells
In order to identify molecular targets of mir-106a,
we used a microRNA database (miRanda) to find potential cell cycle
regulators associated with the inhibition of G1-S cell cycle
progression. We identified cyclin D1, CDK2, CDK6, and
p21CIP1/WAF1 as candidates based on the miRanda
bioinformatics algorithm (Fig. 3E).
Immunoblot assays were performed to investigate possible changes in
the expression of those candidates after transfection with
mir-106a. Interestingly, mir-106a transfection induced a
downregulation in both cyclin D1 and CDK6 in EJ cancer cells
(Fig. 3A). However, cyclin E and
CDK2 levels were unchanged. In addition, among the cell cycle
inhibitors examined, p21CIP1/WAF1 expression was
significantly increased >3-fold over expression in the control
(Fig. 3B). However,
p27KIP1 and p53 were not altered by mir-106a
transfection.
Since cyclin D1/CDK6 forms a complex that
phosphorylates the Rb protein to aid cell cycle progression, we
investigated the kinase activity of CDK6. Cell lysates from the
mir-106a transfectants were immunoprecipitated with a CDK6
antibody, and then GST-pRb was added as the substrate with a kinase
buffer containing radiolabeled 32P-aTP. The kinase
activity of CDK6 was reduced >30% in mir-106a transfectants
compared to the control or transfection with the mir-106a inhibitor
(Fig. 3C). This suggests that
mir-106a clearly targets cyclin D1/CDK6-associated growth signals
in bladder cancer cells. upregulation of p21CIP1/WAF1
expression by mir-106a was again validated in immunoprecipitation
experiments with a CDK6 antibody followed by immunoblotting with an
antibody against p21CIP1/WAF1 or CDK6 (control). Complex
formation of p21CIP1/WAF1 with CDK6 was increased
>4-fold in the mir-106a-transfected cells compared to the
control (Fig. 3D). Taken together,
these results demonstrate that p21CIP1/WAF1 is a
critical effector in mir-106a-mediated G1-phase cell cycle arrest
and growth inhibition.
mir-106a suppresses migration and
invasion of bladder cancer cells
When normal bladder cells are transformed, they
frequently acquire the ability to invade other tissues. We
investigated whether the transfection of mir-106a suppresses the
migration and invasion of bladder cancer cells. In wound-healing
assays, mir-106a transfectants exhibited slower growth into the
wounded area than control cells or cells transfected with the
mir-106a inhibitor (Fig. 4A).
Wound-closure rate was ~60% slower after mir-106a transfection.
Similarly, Boyden chamber assays showed that invasiveness of EJ
bladder cancer cells was reduced >30% after mir-106a
transfection (Fig. 4B). These
results suggest that mir-106a possesses a tumor suppressive
function in the migration and invasion of bladder cancer cells.
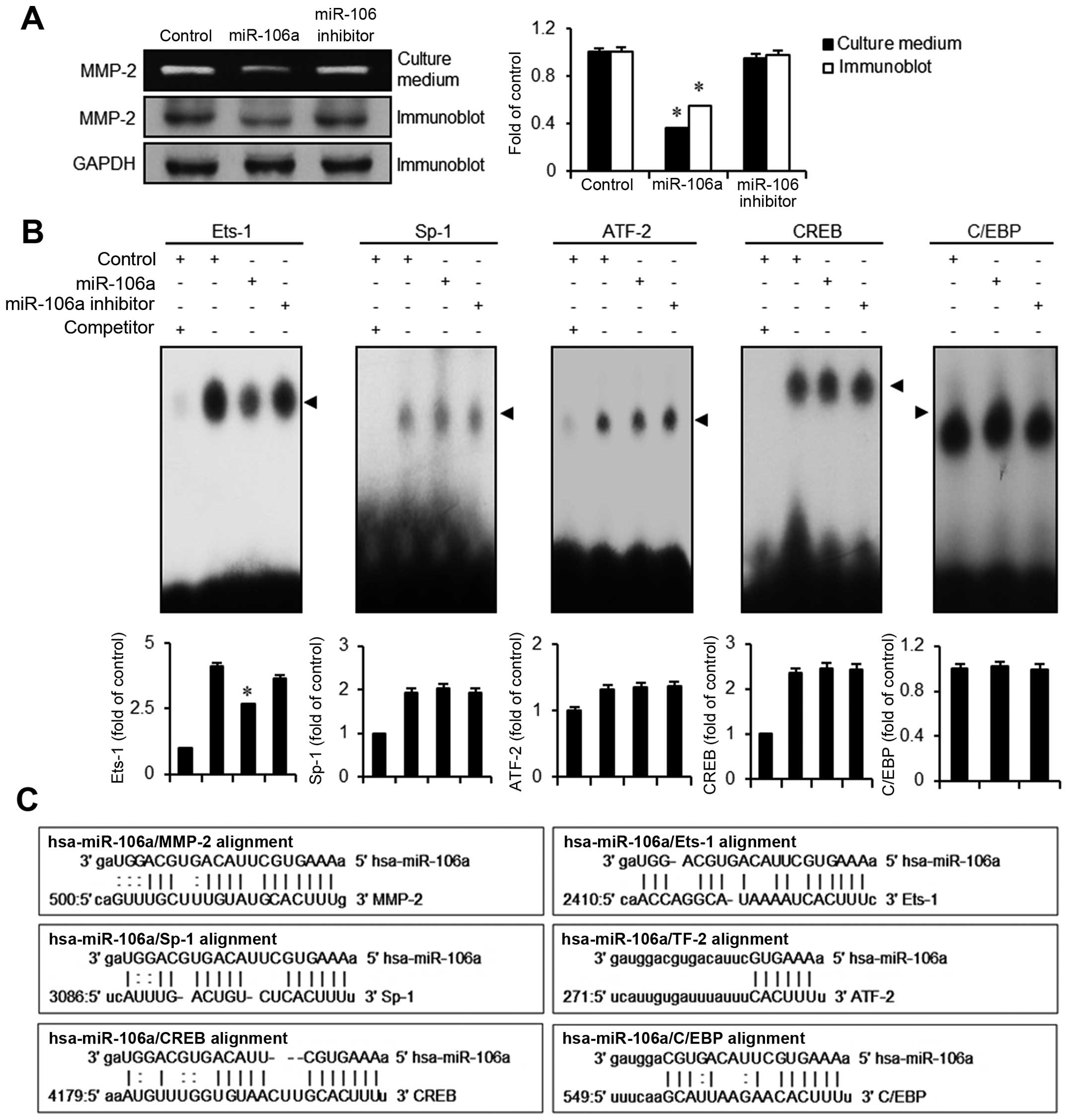
mir-106a downregulates MMP-2 expression
via activation of Ets-1
Transformation of bladder epithelia often includes
the upregulation of proteases such as MMPs to create spaces for the
cancer cells to migrate. using the miRanda sequence a lignment
algorithm for mir-106, we found several putative targets including
MMP-2, Ets-1, Sp-1, ATF-2, CREB, and C/EBP (Fig. 5C). In order to verify whether MMP-2
is modulated by mir-106a transfection, we performed gelatin
zymography assays. In conditioned culture medium obtained from
mir-106a transfectants, gelatinase activity of MMP-2 was
significantly lower (>60%) than in the control. Correspondingly,
in immunoblot assays, MMP-2 protein expression was significantly
lower than that in the control or the mir-106 inhibitor (Fig. 5A). We next investigated which
MMP-2-mediated transcription factors are modulated by mir-106a.
Because Ets-1, Sp-1, ATF-2, CREB, and C/EBP were the candidates
identified from the miRanda DB search, we assessed alterations in
binding activities of these factors in mir-106a transfectants using
EMSA assays. Among transcription factors tested, only Ets-1
exhibited a reduced activity in the binding motif (Fig. 5B). Activities of Sp-1, ATF-2, CREB,
and C/EBP were unchanged by the introduction of mir-106a. These
results suggest that mir-106a abrogates the expression of MMP-2
modulated by the transcription factor Ets-1, resulting in the
inhibition of migration and invasion in bladder cancer EJ
cells.
Discussion
Bladder cancer is one of the most notorious
malignancies in Western countries. The incidence is estimated to be
1 in 26 for males, and ~74,000 new cases were diagnosed in 2015 in
the US (17). In this study, we
report the novel finding that mir-106a regulates the proliferation,
migration, and invasion of bladder cancer cells through modulating
effectors such as early signaling molecules (ERK, p38MAPK, and
JNK), cell cycle regulators (cyclin D1, CDK6, and
p21CIP1/WAF1), and extracellular protein degradation
enzymes (MMP-2 and Ets-1).
Initially, we examined the basal expression of
mir-106a in three bladder carcinoma cell lines. The basal
expression level of mir-106a was significantly lower in cancer cell
lines in comparison to control. The basal expression of mir-106a in
cancers is controversial and seems to be dependent on tumor type
(18–21). For example, Volinia and colleagues
performed miRnome analysis to verify whether the expression of
microRNA correlates with malignancies, and a total of 540 clinical
specimens including lung, breast, stomach, prostate, colon, and
pancreatic cancers were investigated. mir-106a was determined to be
upregulated in colon, pancreas, and prostate cancers. However, in
breast cancers, mir-106a expression was downregulated with respect
to its expression in corresponding normal tissues (22). Controversially, Diaz and coworkers
reported that mir-106a expression was significantly downregulated
in tumor specimens from 110 colorectal cancer patients, and is
correlated with poor survival (P<0.04) (21). In agreement with some previous
results, our results showed lower expression of mir-106a in bladder
cancer cells compared to normal urothelial cells. Cell viability
assays showed a dose-dependent decrease of EJ cancer cell
proliferation after transfection with mir-106a. Interestingly, ERK
phosphorylation was significantly reduced by mir-106a transfection,
whereas phosphorylation of p38 and JNK was upregulated.
Hyperactivation of MAPKs, particularly ERK, or genetic mutations in
the RAS-RAF-MEK-ERK signaling cascade have been reported as
critical events in tumorigenesis of various tissue types. For
example, in melanoma, BRAF mutation at position 600 (V600E) has
been reported as a mutation hot spot or signature mutation
(23–25). Even melanomas with wild-type BRAF
frequently show increased phosphorylation of ERK through autocrine
stimulation with growth factors such as fibroblast growth factor
(FGF) and insulin-like growth factor (IGF) (26,27).
This suggests that cancer cells may be addicted to growth stimuli
from ERK activation. Therefore, it is noteworthy that mir-106a
inhibits ERK signaling in bladder cancers. In addition to the
inhibition of phospho-ERK signaling, activation of stress-activated
protein kinases p38 and JNK (28)
was observed in mir-106a transfectants. These results suggest that
mir-106a exerts an antiproliferative effect on bladder cancer cells
through modulating the MAPK signaling pathway.
Cell cycle analysis showed that mir-106a-transfected
bladder cancer cells accumulated in G1-phase, suggesting that
cyclins might be associated with mir-106a-mediated cell cycle
arrest. In bladder cancers, cyclin D1 and E were reported to be key
regulators associated with disease initiation and progression
(29,30). We performed an immunoblot assay and
a kinase complex assay to verify which cyclin participated in the
mir-106a signaling pathways. Interestingly, only cyclin D1 was
shown to be modulated by mir-106a. This result was supported by the
observation that the activity of its corresponding kinase CDK6 was
equally diminished by transfection with mir-106a. As a matter of
fact, cumulative results have demonstrated that a 6–7 base seed
sequence was sufficient for miRNA regulation (31,32).
Therefore, we employed the 6 base sequence of CDK2 to know whether
mir-106a regulate the CDK2 level in bladder cancer cells. In this
study our results indicated that 6 base seed sequence of CDK2 did
not influence the miRNA regulation. This result suggested that CDK2
was not affected by the introduction of mir-106a in bladder cancer
cells. We next investigated whether CDK inhibitors were
correspondingly altered by mir-106a transfection. Among CDK
inhibitors, p21CIP1/WAF1 was significantly upregulated
in mir-106a transfectants. This was further verified by
immunoprecipitation assays using a CDK6 antibody followed by
immunoblotting with a p21CIP1/WAF1 antibody. A recent
study demonstrated that CDK6 might be an effective target of
mir-29c for suppressing bladder cancer progression (33). In agreement with those results, our
data showed that mir-106a inhibited cyclin D1 and CDK6 expression
in bladder cancer cells. This study demonstrated that mir-106a
inhibits proliferation of bladder cancer EJ cells by targeting the
p21CIP1/WAF1/cyclin D1/CDK6 cell cycle cascade.
Previous studies have shown that MMP-2 upregulation
in bladder cancer is a key step in migration and invasion (6,13,16).
MicroRNAs, including mir-430 and mir-20b, are known to be capable
of regulating migration and invasion by coordinating MMP-2
expression (13,34). In this study, mir-106a
overexpression blocked the migration and invasion of bladder cancer
cells. The inhibitory effect was in part due to the inhibition of
MMP-2 activity as verified by gelatin zymography assays and
immunoblotting. MMP-2 expression in bladder cancers is tightly
regulated by transcription factors including p53, AP-1, Ets-1,
C/EBP, CREB, PEA3, Sp1, ATF2, and AP-2 (35). In this study, Ets-1 was identified
as an essential transcription factor responsible for the
mir-106a-mediated MMP-2 inhibition in bladder cancer cells. Our
results show that mir-106a may suppress migration and invasion by
downregulating Ets-1 and MMP-2 expression in bladder cancer
cells.
In conclusion, we verified that the expression level
of mir-106a is abnormally downregulated in bladder cancer cells.
Introduction of mir-106a impaired the proliferation of bladder
cancer EJ cells through modulating MAPK (ERK/p38/JNK) signaling and
cell cycle regulators (p21CIP1/WAF1/cyclin D1/CDK6). In
addition, mir-106a overexpression impeded MMP-2 expression by
inactivating the Ets-1 binding motif, which led to a reduction in
the migration and invasion of bladder cancer cells. Therefore, our
data suggest that mir-106a might serve as a potential target for
treating bladder malignancies. Furthermore, our results indicate
that bioinformatics might be a useful tool for predicting the
targets of miRs involved in the regulation of tumor cells.
Acknowledgments
This study was supported by the National Research
Foundation of Korea (NRF) grant by the government of Korea (MSIP)
(no. 2014007036) and the Functional Districts of the Science Belt
Support Program, Ministry of Science, ICT and Future Planning (no.
2015K000284).
References
|
1
|
Hayes J, Peruzzi PP and Lawler S:
MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol
Med. 20:460–469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Calin GA, Dumitru CD, Shimizu M, Bichi R,
Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al:
Frequent deletions and down-regulation of micro-RNA genes miR15 and
miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci
USA. 99:15524–15529. 2002. View Article : Google Scholar
|
|
3
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koturbash I, Zemp FJ, Pogribny I and
Kovalchuk O: Small molecules with big effects: The role of the
microRNAome in cancer and carcinogenesis. Mutat Res. 722:94–105.
2011. View Article : Google Scholar
|
|
5
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar
|
|
6
|
Eissa S, Ali-Labib R, Swellam M, Bassiony
M, Tash F and El-Zayat TM: Noninvasive diagnosis of bladder cancer
by detection of matrix metalloproteinases (MMP-2 and MMP-9) and
their inhibitor (TIMP-2) in urine. Eururol. 52:1388–1396. 2007.
|
|
7
|
Jiang X, Du L, Wang L, Li J, Liu Y, Zheng
G, Qu A, Zhang X, Pan H, Yang Y, et al: Serum microRNA expression
signatures identified from genome-wide microRNA profiling serve as
novel noninvasive biomarkers for diagnosis and recurrence of
bladder cancer. Int J Cancer. 136:854–862. 2015. View Article : Google Scholar
|
|
8
|
Ratert N, Meyer HA, Jung M, Lioudmer P,
Mollenkopf HJ, Wagner I, Miller K, Kilic E, Erbersdobler A, Weikert
S, et al: miRNA profiling identifies candidate mirnas for bladder
cancer diagnosis and clinical outcome. J Mol Diagn. 15:695–705.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Puerta-Gil P, García-Baquero R, Jia AY,
Ocaña S, Alvarez-Múgica M, Alvarez-Ossorio JL, Cordon-Cardo C, Cava
F and Sánchez-Carbayo M: miR-143, miR-222, and miR-452 are useful
as tumor stratification and noninvasive diagnostic biomarkers for
bladder cancer. Am J Pathol. 180:1808–1815. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang XL, Xie HY, Zhu CD, Zhu XF, Cao GX,
Chen XH and Xu HF: Increased miR-141 expression is associated with
diagnosis and favorable prognosis of patients with bladder cancer.
Tumour Biol. 36:877–883. 2015. View Article : Google Scholar
|
|
11
|
Rouanne M, Loriot Y, Lebret T and Soria
JC: Novel therapeutic targets in advanced urothelial carcinoma.
Crit Rev Oncol Hematol. 98:106–115. 2016. View Article : Google Scholar
|
|
12
|
Iyer G, Al-Ahmadie H, Schultz N, Hanrahan
AJ, Ostrovnaya I, Balar AV, Kim PH, Lin O, Weinhold N, Sander C, et
al: Prevalence and co-occurrence of actionable genomic alterations
in high-grade bladder cancer. J Clin Oncol. 31:3133–3140. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park SL, Cho TM, Won SY, Song JH, Noh DH,
Kim WJ and Moon SK: MicroRNA-20b inhibits the proliferation,
migration and invasion of bladder cancer EJ cells via the targeting
of cell cycle regulation and Sp-1-mediated MMP-2 expression. Oncol
Rep. 34:1605–1612. 2015.PubMed/NCBI
|
|
14
|
Yun SJ, Jeong P, Kim WT, Kim TH, Lee YS,
Song PH, Choi YH, Kim IY, Moon SK and Kim WJ: Cell-free microRNAs
in urine as diagnostic and prognostic biomarkers of bladder cancer.
Int J Oncol. 41:1871–1878. 2012.PubMed/NCBI
|
|
15
|
Moon SK, Jung SY, Choi YH, Lee YC,
Patterson C and Kim CH: PDTC, metal chelating compound, induces G1
phase cell cycle arrest in vascular smooth muscle cells through
inducing p21Cip1 expression: Involvement of p38 mitogen activated
protein kinase. J Cell Physiol. 198:310–323. 2004. View Article : Google Scholar
|
|
16
|
Lee SJ, Cho SC, Lee EJ, Kim S, Lee SB, Lim
JH, Choi YH, Kim WJ and Moon SK: Interleukin-20 promotes migration
of bladder cancer cells through extracellular signal-regulated
kinase (ERK)-mediated MMP-9 protein expression leading to nuclear
factor (NF-χB) activation by inducing the up-regulation of
p21(WAF1) protein expression. J Biol Chem. 288:5539–5552. 2013.
View Article : Google Scholar
|
|
17
|
American Cancer Society: Cancer Facts and
Figures 2015. American Cancer Society; Atlanta, GA: 2015,
http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdfurisimplewww.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf.
|
|
18
|
Wang F, Zheng Z, Guo J and Ding X:
Correlation and quantitation of microRNA aberrant expression in
tissues and sera from patients with breast tumor. Gynecol Oncol.
119:586–593. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Feng B, Dong TT, Wang LL, Zhou HM, Zhao
HC, Dong F and Zheng MH: Colorectal cancer migration and invasion
initiated by microRNA-106a. PLoS One. 7:e434522012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Z, Liu M, Zhu H, Zhang W, He S, Hu C,
Quan L, Bai J and Xu N: miR-106a is frequently upregulated in
gastric cancer and inhibits the extrinsic apoptotic pathway by
targeting FAS. Mol Carcinog. 52:634–646. 2013. View Article : Google Scholar
|
|
21
|
Díaz R, Silva J, García JM, Lorenzo Y,
García V, Peña C, Rodríguez R, Muñoz C, García F, Bonilla F, et al:
Deregulated expression of miR-106a predicts survival in human colon
cancer patients. Genes Chromosomes Cancer. 47:794–802. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hodis E, Watson IR, Kryukov GV, Arold ST,
Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C,
et al: A landscape of driver mutations in melanoma. Cell.
150:251–263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhuang L, Lee CS, Scolyer RA, McCarthy SW,
Palmer AA, Zhang XD, Thompson JF, Bron LP and Hersey P: Activation
of the extracellular signal regulated kinase (ERK) pathway in human
melanoma. J Clin Pathol. 58:1163–1169. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smalley KS: A pivotal role for ERK in the
oncogenic behaviour of malignant melanoma? Int J Cancer.
104:527–532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yayon A, Ma YS, Safran M, Klagsbrun M and
Halaban R: Suppression of autocrine cell proliferation and
tumorigenesis of human melanoma cells and fibroblast growth factor
transformed fibroblasts by a kinase-deficient FGF receptor 1:
Evidence for the involvement of Src-family kinases. Oncogene.
14:2999–3009. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu GS: Role of mitogen-activated protein
kinase phosphatases (MKPs) in cancer. Cancer Metastasis Rev.
26:579–585. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eissa S, Ahmed MI, Said H, Zaghlool A and
El-Ahmady O: Cell cycle regulators in bladder cancer: Relationship
to schistosomiasis. IUBMB Life. 56:557–564. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shan G and Tang T: Expression of cyclin D1
and cyclin E in urothelial bladder carcinoma detected in tissue
chips using a quantum dot immunofluorescence technique. Oncol Lett.
10:1271–1276. 2015.PubMed/NCBI
|
|
31
|
Zang WQ, Yang X, Wang T, Wang YY, Du YW,
Chen XN, Li M and Zhao GQ: MiR-451 inhibits proliferation of
esophageal carcinoma cell line EC9706 by targeting CDKN2D and
MAP3K1. World J Gastroenterol. 21:5867–5876. 2015.PubMed/NCBI
|
|
32
|
Takahashi Y, Forrest AR, Maeno E,
Hashimoto T, Daub CO and Yasuda J: MiR-107 and MiR-185 can induce
cell cycle arrest in human non small cell lung cancer cell lines.
PLoS One. 4:e66772009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao X, Li J, Huang S, Wan X, Luo H and Wu
D: MiRNA-29c regulates cell growth and invasion by targeting CDK6
in bladder cancer. Am J Transl Res. 7:1382–1389. 2015.PubMed/NCBI
|
|
34
|
Liu L, Zhao X, Zhu X, Zhong Z, Xu R, Wang
Z, Cao J and Hou Y: Decreased expression of miR-430 promotes the
development of bladder cancer via the upregulation of CXCR7. Mol
Med Rep. 8:140–146. 2013.PubMed/NCBI
|
|
35
|
Qin H, Sun Y and Benveniste EN: The
transcription factors Sp1, Sp3, and AP-2 are required for
constitutive matrix metalloproteinase-2 gene expression in
astroglioma cells. J Biol Chem. 274:29130–29137. 1999. View Article : Google Scholar : PubMed/NCBI
|