Introduction
Lysine-specific demethylase 1 (LSD1/KDM1) was the
first discovered histone demethylase that removed the mono-methyl
and di-methyl moieties from H3-K4 via a flavin adenine dinucleotide
(FAD)-dependent monoamine oxidoreductase and governed transcription
regulation by serving as an epigenetic coregulator (1,2). In
previous studies, the function of LSD1 appeared to be as a tumor
suppressor or oncogene depending on the tumor type. For example,
overexpression of LSD1 was noted in neuroblastoma (3), non-small cell lung cancer (4), colorectal cancer (5) and prostate cancer (6) and was found to promote proliferation,
migration and invasion. While in breast cancer, LSD1 was
downregulated and inhibited tumor metastasis by the suppression of
TGFβ (7,8). Thus, it is reasonable that the
function of LSD1 is opposing due to the distinct target genes in
different types of cancer. As reported, inhibition of HDACs and
LSD1 are potential combination therapies in glioblastoma multiforme
(GBM) (9), but the function and
post-transcription of LSD1 remain unclear.
Deubiquitylating enzymes (DUBs) are a number of
proteins that antagonize ubiquitylation by cleaving polyubiquitin
or monoubiquitin and thus afford the stabilization or transcription
regulation of the substrates. Ubiquitin specific peptidase 7
(USP7), also known as human herpesvirus-associated
ubiquitin-specific protease (HAUSP) is a member of the DUB family
and was first revealed to deubiquitinate and stabilize p53
(10,11). Subsequently, numerous proteins have
been reported as potential substrate/binding partners of USP7, such
as Epstein-Barr nuclear antigen 1 (EBNA1) (12), PTEN (13), INK4a (14), the transcription factors FOXO4
(15) and REST (16). USP7 is involved in the cell cycle by
the regulation of many proteins, as well as tumor suppressors or
oncogenes. However, the expression pattern and substrates for USP7
in glioma patients are still unclear.
Glioma is the most common primary aggressive
malignant brain tumor, among which, GBM is well known as the
highest grade glioma and one of the most lethal forms of cancer in
humans (17). Glioblastoma therapy,
over the past decade, which combines surgery, postoperative
chemotherapy and radiation therapy has failed to benefit all
patients suffering from disease progression, equally. The overall
median survival is still less than 15 months (18,19),
thus individualized therapy such as targeting several aberrant
epigenetic functions should be considered as a potentially valuable
approach for glioma patients.
Materials and methods
Patients and tissue samples
The study was approved by the Research Ethics
Committee of Central South University. Human brain samples were
obtained from 150 patients who underwent surgical treatment at the
Second Xiangya Hospital of Central South University, Changsha,
China. Written informed consent was obtained from all the patients.
The fresh glioma specimens were obtained between January 2008 and
December 2012. The patients who had received radiotherapy or
chemotherapy prior to surgery were excluded. Sections of the
specimen were snap frozen and stored at −80°C for mRNA isolation;
other sections of the specimen were used for histological
sectioning. Additionally, 10 cases of normal brain tissue samples
were obtained from patients who underwent surgery for decompression
treatment due to severe head injuries other than malignancies. All
the glioma samples were verified by the World Health Organization
(WHO) 2007 classification standard. The details of the patients are
presented in Table I. In the
follow-up period, overall survival was calculated from diagnosis to
death; the total follow-up period was 60 months.
 | Table IMean values of LSD1 mRNA expression in
the clinical glioma samples and normal control tissues, and
comparison with clinicopathological variables. |
Table I
Mean values of LSD1 mRNA expression in
the clinical glioma samples and normal control tissues, and
comparison with clinicopathological variables.
| Variables | n | LSD1 expression (mean
± SD) | P-value |
|---|
| Tissue type | | | <0.05 |
| Glioma | 150 | 6.973±0.454 | |
| Normal control | 10 | 1.342±0.239 | |
| Gender | | | >0.05 |
| Male | 86 | 6.731±0.872 | |
| Female | 64 | 6.422±0.764 | |
| Age (years) | | | >0.05 |
| <60 | 84 | 6.453±0.784 | |
| ≥60 | 66 | 6.891±0.545 | |
| KPS | | | <0.05 |
| <80 | 73 | 6.584±0.713 | |
| ≥80 | 77 | 6.136±0.376 | |
| WHO grade | | | <0.05 |
| I | 28 | 3.741±0.131 | |
| II | 29 | 5.349±0.394 | |
| III | 45 | 7.032±0.579 | |
| IV | 48 | 8.712±0.732 | |
Cell culture and transfection
The human A172 and T98G GBM cells were obtained from
the American Type Culture Collection (ATCC; Manassas, VA, USA). The
aforementioned cells were cultured in Dulbecco's modified Eagle's
medium (DMEM) with 10% fetal bovine serum (FBS) (Gibco, Los
Angeles, CA, USA) and incubated at 37°C in humidified 5%
CO2. When the cells were grown to 70% confluence,
relative plasmids or siRNAs were transfected into the cells using
Lipofectamine 2000 reagent (Invitrogen Life Technologies, Carlsbad,
CA, USA) according to the manufacturer's instructions. The siRNAs
and the negative controls were synthesized by Shanghai GenePharma
(Shanghai, China). The knockdown efficiency was measured 48 h after
transfection.
Real-time PCR
Total RNA was extracted from the glioma specimens or
the control normal brain tissues or the GBM cells using TRIzol
reagent (Invitrogen Life Technologies). The concentration and
purity of RNA were determined, and then ~2 µg RNA was used
to synthesize cDNA using the cDNA Reverse Transcription kit
(TransGen Biotech, Inc., Beijing, China). Real-time PCR reaction
amplification was performed using the SYBR-Green PCR Master Mix on
a 7500 Fast Real-Time PCR system cycler (Applied Biosystems, Foster
City, CA, USA). The expression of the relative genes was analyzed
using the 2−ΔΔCt method. GAPDH mRNA was used as an
internal control to normalize the selected genes in the same
sample. The amplification protocol consisted of denaturation at
98°C for 5 min, followed by 40 cycles of denaturation at 98°C for
10 sec and annealing and extension at 60°C for 30 sec. The process
was repeated for 40 cycles.
Co-immunoprecipitation assay
For co-immunoprecipitation assay (co-IP), the cells
were washed with cold phosphate-buffered saline (PBS) and lysed
with cold lysis buffer (50 mM Tris-HCl pH 7.4, 0.5% SDS, 150 mM
NaCl, 1% NP-40, and 1 mM EDTA) at 4°C for 45 min, followed by
centrifugation at 132,000 rpm for 15 min at 4°C. The supernatant
was collected and incubated with appropriate primary antibodies or
normal rabbit/mouse immunoglobin G (IgG) as a negative control, on
a rotator overnight at 4°C. After being incubated with protein A/G
Sepharose CL-4B beads for 2 h at 4°C, the beads were washed 5 times
with lysis buffer and the immune complexes were subjected onto
SDS-PAGE, followed by western blotting detection.
GST pull-down assay
The GST fusion construct was expressed in BL21
Escherichia coli cells, and the in vitro
transcription and translation experiments were performed using
rabbit reticulocyte lysate (TNT systems; Promega) according to the
manufacturer's recommendation. In the GST pull-down assay, 10
µg of GST fusion protein was incubated with 10 µl of
the in vitro transcribed/translated products at room
temperature for 30 min. Thirty microliters of glutathione-Sepharose
beads was then added to the binding reaction and mixed at 4°C for 2
h. After being washed three times with binding buffer, the immune
complexes were resolved by SDS-PAGE, followed by western blot
detection.
Western blotting
Total proteins were purified from the GBM cells and
equal amount of protein lysate (30 µg) was separated on an
8% polyacrylamide gel (Invitrogen Life Technologies). Then the gel
was transferred onto a PVDF membrane at 400 mA for 1 h. After being
blocked with 5% non-fat milk, the membrane was incubated with the
primary antibody against USP7 (sc-30164, 1:1,000; Santa Cruz
Biotechnology, Inc.) or LSD1 (ab17721, 1:1,000; Abcam) or actin
(1:2,000; Beyotime Institute of Biotechnology) overnight at 4°C.
Finally, the membrane was incubated with horseradish
peroxidase-conjugated goat anti-rabbit or goat anti-mouse antibody
at room temperature for 1 h. The ECL detection system (BeyoECL
Plus; Beyotime Institute of Biotechnology) was used to visualize
the immunoblots.
Cell cycle analysis
The cell cycle was analyzed by flow cytometry (FCM).
GBM cells were synchronized by serum starvation for 24 h and
released with 10% FBS for an appropriate period of time, and then
the cells were harvested, washed with PBS, and fixed with 70%
ethanol at 4°C overnight. Cells were incubated with RNase
(Sigma-Aldrich) in PBS for 30 min at 37°C for 30 min. Cell cycle
data were collected with FACSCalibur flow cytometer
(Becton-Dickinson) and analyzed with FlowJo 7.6 software. Each
experiment was performed in triplicate.
Cell proliferation analysis
The cell proliferation analysis was performed using
the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) assay. The cells were plated into a 12-well culture plate
(10,000 cells/well), and incubated in a humidified atmosphere of 5%
CO2 at 37°C. At 24, 48, 72 or 96 h, 10 µl was
added to each well (MTT; Sigma-Aldrich), and then incubation was
carried out at 37°C for 4 h. Dimethyl sulfoxide (DMSO)
(Sigma-Aldrich) was used to terminate the reaction. The optical
absorbance was measured using a micro-plate reader (Bio-Rad,
Hercules, CA, USA) at 570 nm and quantification of the cell
viability was determined according to the optical density
values.
EdU cell proliferation assay
Cells were seeded onto 24-well plates for 24 h and
transfected with the indicated siRNAs or plasmids using
Lipofectamine 2000 (Invitrogen Life Technologies). Cell
proliferation ELISA EdU kits (Roche Applied Science, Indianapolis,
IN, USA) were used according to the manufacturer's instructions.
Forty-eight hours after transfection, 5-ethynyl-2′-deoxyuridine
(EdU) was added to the medium for an additional 4 h at 37°C, and
then the wells were washed 3 times with washing solution (200
µl/well). Four percent formaldehyde was added to the cells
for 30 min, and 2 mg/ml glycine was incubated for 5 min. Finally,
4′,6-diamidino-2-phenylindole (DAPI) was used to stain the nuclei.
The experiments were performed in triplicate.
Transwell assay
A Transwell chamber was precoated with 6 µl
Matrigel at 4°C overnight and 5×104 cells were seeded
into 500 µl serum-free DMEM on the upper chamber. DMEM
supplemented with 10% FBS was added to the lower chamber. After
being allowed to invade for 10 h, the invading cells on the lower
surface were fixed with 70% ethanol followed by crystal violet
staining, while the remaining cells on the upper chamber were
removed using cotton swabs. The number of invaded cells was
calculated under a microscope in four random fields of vision.
Statistical analysis
SPSS software 17.0 (SPSS Inc., Chicago, IL, USA) was
used for statis tical analysis. The significance between groups was
analyzed using one-way analysis of variance (ANOVA) and Student's
t-tests. A Kaplan-Meier plot and univariate Cox regression analysis
were used to analyze the glioma patient morbidity. Log-rank
analysis was used to test the differences between groups. The means
± standard deviation was calculated for all experiments. P<0.05
was considered to be statistically significance;
*P<0.05, **P<0.01 are indicated in the
figures and legends.
Results
LSD1 is identified as a USP7-interacting
protein
In order to investigate the potential substrates of
USP7 in glioblastoma, we first used co-IP to identify the
USP7-interacting proteins in A172 cells, as shown in Fig. 1A, firstly, by immunoprecipitation
with antibodies against USP7, and then followed by western blotting
against LSD1 antibodies. The data indicated that USP7 was
associated with LSD1 in vivo, but not with the IgG control.
Reciprocal immunoprecipitation with anti-LSD1 and immunoblotting
with anti-USP7 also indicated this fact. This interaction was also
confirmed with endogenous proteins in the T98G cells (Fig. 1B) Notably, this interaction was
specific, since USP7 did not associate with another lysine-specific
demethylase 2 (LSD2) (data not shown).
To investigate whether USP7 interacts with LSD1
directly, a GST pull-down experiment was performed. Glutathione
S-transferase fusion protein (GST-zIP) was immobilized on
glutathione Sepharose 4B beads, and subsequently incubated with
in vitro transcribed/translated FLAG-tagged LSD1, as shown
in Fig. 1C. The GST-USP7 fusion
protein was found to bind with LSD1 directly in vitro.
Collectively, these experiments supported our observation that USP7
is physically associated with LSD1 in vivo and in
vitro.
USP7 inhibits LSD1 ubiquitination and
stabilizes LSD1 in vivo
Since USP7 was shown to deubiquitinate and stabilize
p53, MDM2 and SIRT1, it was also feasible that USP7 may regulate
LSD1 function via its deubiquitinase activity. Firstly, A172 cells
as well as T98G cells were infected with a control siRNA, siUSP7#1,
or siUSP7#2 and the knockdown efficiency was detected. As shown in
Fig. 2A, the USP7 mRNA level was
significantly reduced when cells were infected with siUSP7#1 or
siUSP7#2 as compared with the level in the control siRNA group.
Concomitantly, the protein level, as detected by western blotting
revealed that USP7 expression was also greatly reduced in the siRNA
groups than that noted in the control group and siUSP7#2 was more
effective and used for further experiments. Furthermore, after USP7
was knocked down, we investigated the expression of LSD1 and as
expected, although the mRNA level was not changed (Fig. 2B, left panels), the protein level of
LSD1 was dramatically reduced (Fig.
2B, right panels). This indicated that USP7 may influence LSD1
through post-transcription modification. Consistently, we
overexpressed wild-type USP7 plasmid or catalytically inactive USP7
(USP7/C223A) mutant plasmid in the A172 and T98G cells and it was
revealed that the protein level of LSD1 only increased in the
wt-USP7-transfected groups, but not in the catalytically inactive
USP7 (USP7/C223A) mutant group (Fig.
2C), where there was no marked change in the mRNA level.
To determine the effect of USP7 on LSD1 protein
ubiquitination, USP7 was knocked down by siRNA in the A172 and T98G
cells, and the cells were harvested after MG132 (a
proteasome-specific inhibitor) treatment. We found that MG132 was
successful in rescuing the LSD1 protein but not mRNA from the
degradation in the USP7-knockdown cells (Fig. 2D). Based on the aforementioned
experiments, we demonstrated that USP7 could stabilize LSD1
possibly based on its deubiquitinating activity.
LSD1 plays a critical role in the
proliferation and invasion of glioblastoma cells
As reported, LSD1 is overexpressed in glioblastoma
(9), but the function of LSD1
requires further exploration. Thus, to investigate the function,
two specific siRNAs targeting LSD1 were successfully developed and
transfected into the A172 and T98G cells. The knockdown efficiency
was nearly 90%, and as shown in Fig.
3A, siLSD1#2 was more effective and was used for further
experimentation. As shown in Fig.
3B, the proliferation of the A172 and T98G cells as determined
by MTT assay was significantly suppressed after treatment with
siLSD1 compared with the control group of cells. The result were
corroborated using the EdU proliferation assay (Fig. 3C). In the cell cycle distribution
assay, knockdown of LSD1 caused G0/G1 arrest compared with the
control group (Fig. 3D). In the
Transwell assay, there were less cells that crossed the membranes
in the LSD1-knockdown group than that noted in the control group
(Fig. 3E). The aforementioned
results illustrated that the downregulation of LSD1 resulted in the
inhibition of glioblastoma cell proliferation and invasion.
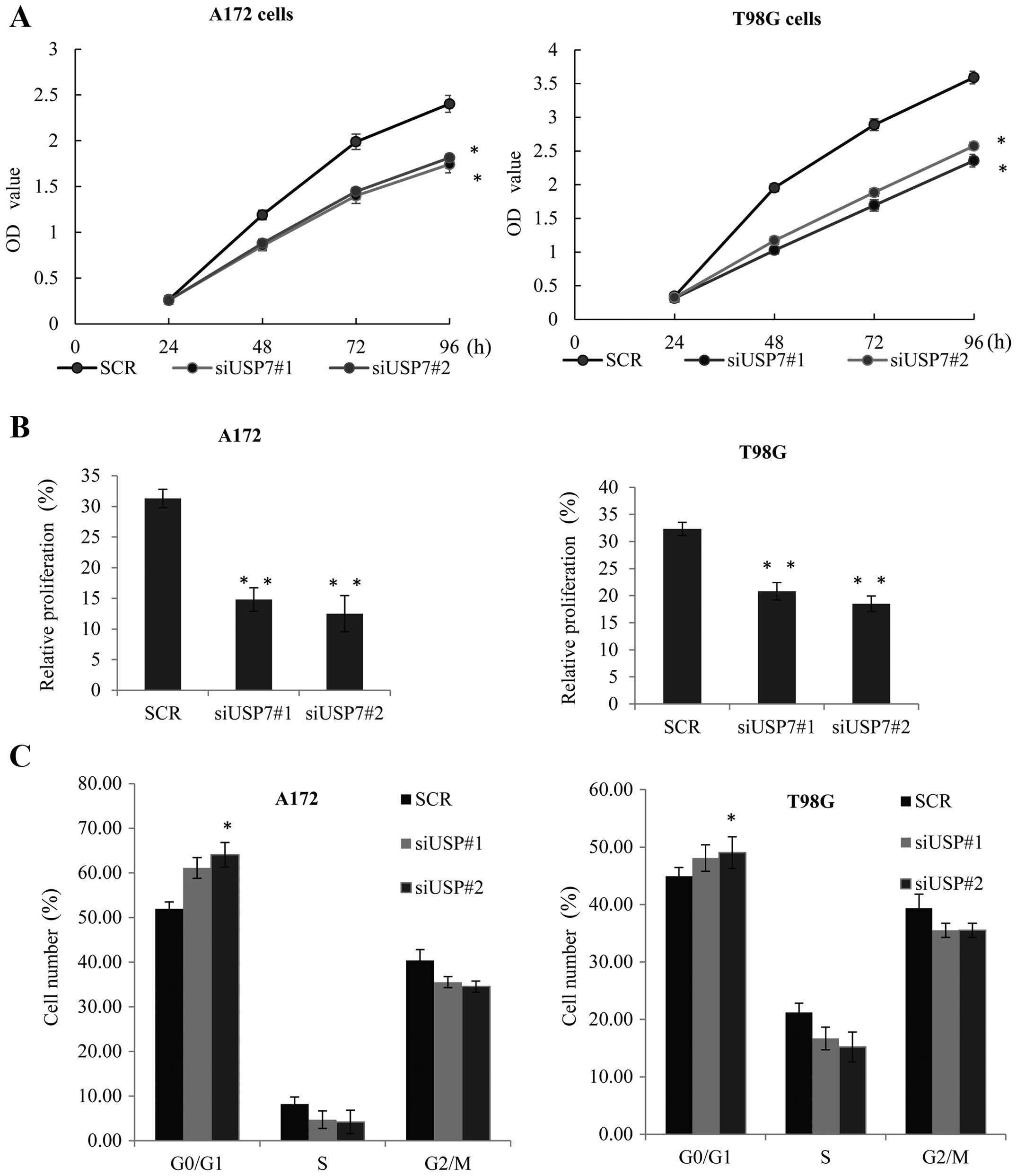
USP7 promotes glioblastoma cell
tumorigenesis by regulation of LSD1
In order to evaluate the effect of USP7 on
glioblastoma tumorigenesis, USP7 was knocked down in the A172
(Fig. 4A, left panel) and T98G
cells (Fig. 4A, right panel) An MTT
assay was performed. When compared with the control siRNA groups,
the knockdown of USP7 effectively inhibited the cell growth rate.
Similarly, EdU cell proliferation assay was performed in the
aforementioned two cell lines (Fig.
4B), and also demonstrated that knockdown of USP7 reduced their
proliferative ability.
The cell cycle distribution by FCM confirmed that in
the A172 and T98G cells, compared with the control cells, knockdown
of USP7 caused G0/G1 arrest (Fig.
4C), and inhibited cell growth. These observations strongly
suggested that USP7 may play a vital role in promoting GBM cell
proliferation in vitro. We examined the role of LSD1 in the
USP7-mediated GBM cell tumorigenesis using the MTT assay. The
inhibiton of cell proliferation resulting from knockdown of USP7
was partially affected by LSD1 overepression in the A172 and T98G
cells (Fig. 4D). Furthermore, upon
treatment with siUSP7, EdU proliferation assay indicated that the
inhibition of proliferation by USP7 knockdown in the A172 and T98G
cells was markedly augmented by LSD1 overexpression (Fig. 4E). Collectively, these results
indicated that LSD1 had an obviously impact on the ability of USP7
to promote cell proliferation.
USP7 affects glioblastoma cell
invasiveness through stabilization of LSD1
The Transwell assay was used to investigate the
impact of USP7 expression on GBM cell invasion. An equal amount of
A172 control or A172 siUSP7 cells were placed into the Transwell
chamber and as shown in Fig. 5A,
the knockdown of USP7 cells exhibited poor invasive ability
(P<0.05). A similar tendency was also observed between the
T98G-siUSP7 and T98G-control cells (Fig. 5B). These data indicated that USP7
increased the invasive ability of GBM cells.
To confirm whether LSD1 is involved in
USP7-triggered glioblastoma cell invasiveness, LSD1 was
overexpressed in the siUSP7#2-transfected A172 cells, and a
Transwell assay was performed. The effect of USP7 was reduced, as
shown in Fig. 5C. Furthermore, we
created a USP7-knockdown T98G cell line, and a similar tendency was
also observed with ectopic expression of LSD1 (Fig. 5D). These results revealed that
knockdown of USP7 inhibited glioblastoma cell invasiveness through
the regulation of LSD1.
Suppression of the p53 signaling pathway
is involved in the USP7-LSD1 regulated glioblastoma cell
tumorigenesis and metastasis
To investigate the potential downstream effectors
which are regulated by USP7 and LSD1, we next examined several
signaling pathways that contributed to cancer cell proliferation
and invasion, such as the p53, the AKT and the Bcl2 apoptosis
pathways. As shown in Fig. 6A, in
the LSD1-knockdown A172 and T98G cells, the mRNA expression of p53
increased while the mRNA expression of Akt1 and Bcl2 were decreased
when compared to the control group cells. Using western blotting
(Fig. 6B), we confirmed that the
protein expression of p53 was higher in the A172-siLSD1 or the
T98G-siLSD1 cells compared with the vector, while the expression of
Akt1 and Bcl2 presented the opposite tendency. Consistently, in
USP7 knockdown in the A172 cells or T98G cells, the mRNA (Fig. 6C) and protein expression (Fig. 6D) of p53, Akt1 and Bcl2 revealed
similar results. Considering the transcription repression function
of LSD1, it is reasonable to make the hypothesis that LSD1 could
suppress p53 directly. These results suggested that USP7 and p53
are upstream and downstream regulators of LSD1-mediated
proliferation and invasion.
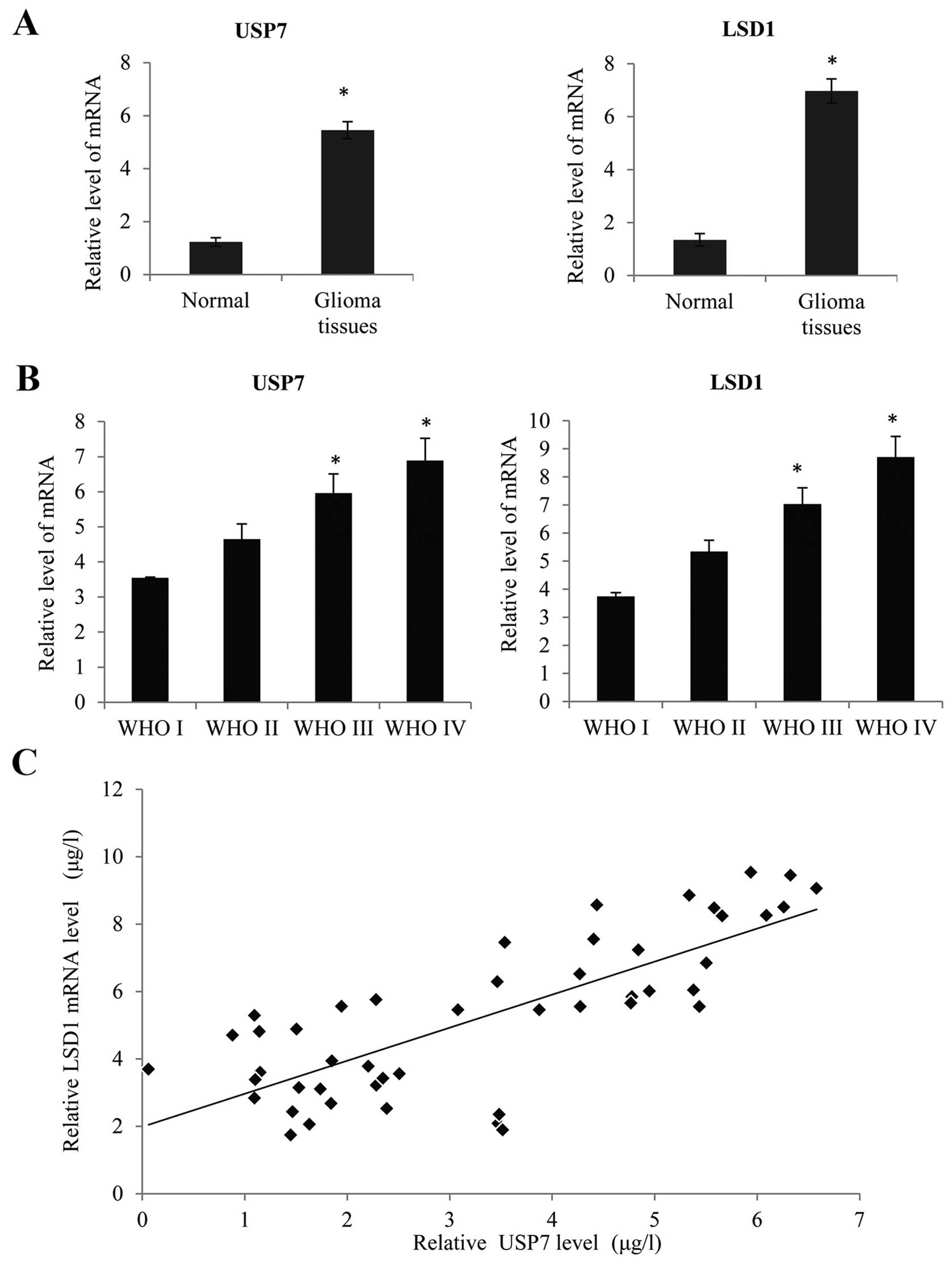
USP7 and LSD1 are frequently upregulated
in glioblastoma and positively correlated with poor GBM
prognosis
Using quantitative real-time PCR (qRT-PCR), we
determined the LSD1 and USP7 mRNA expression, normalized to GAPDH.
As shown in Fig. 7A, we found that
there was no obvious increase in the LSD1 and USP7 expression in
150 glioma tissues compared with non-brain tumor tissues
(P<0.05). There were 57 low-grade (WHO grades I and II) and 103
high-grade tumors (WHO grades III and IV) (Tables I and II). Based on the WHO grade, we found that
LSD1 and USP7 mRNA expression increased notably with the
advancement of WHO grade I to IV (Fig.
7B, P<0.05). Meanwhile, there was a significant positive
correlation between the LSD1 expression and USP7 expression in the
high-grade GBC tissues (WHO grade IV glioblastoma) (Fig. 7C, R=0.06, P<0.05). Furthermore,
according to the details of the follow-up, patients with a higher
LSD1 expression had a worse overall survival after surgery
(Fig. 7D, P<0.05), and a similar
tendency was also observed in patients with a higher USP7
expression (Fig. 7E, P<0.05).
These results suggested that LSD1 and USP7 are upregulated in GBC
tissues and associated with poor prognosis.
| Table IIMean values of USP7 mRNA expression
in clinical glioma samples and comparison with clinicopathological
variables. |
Table II
Mean values of USP7 mRNA expression
in clinical glioma samples and comparison with clinicopathological
variables.
| Variables | n | USP7 expression
(mean ± SD) | P-value |
|---|
| Tissue type | | | <0.05 |
| Glioma | 150 | 5.453±0.324 | |
| Normal
control | 10 | 1.231±0.159 | |
| Gender | | | >0.05 |
| Male | 86 | 5.671±0.592 | |
| Female | 64 | 5.223±0.743 | |
| Age (years) | | | >0.05 |
| <50 | 84 | 5.243±0.954 | |
| ≥50 | 66 | 5.784±0.375 | |
| KPS | | | <0.05 |
| <80 | 73 | 5.754±0.518 | |
| ≥80 | 77 | 5.232±0.459 | |
| WHO grade | | | <0.05 |
| I | 28 | 3.543±0.231 | |
| II | 29 | 4.649±0.434 | |
| III | 45 | 5.958±0.549 | |
| IV | 48 | 6.894±0.632 | |
Discussion
A number of studies have reported that USP7 may
regulate several polyubiquitinated substrates by its
deubiquitinating activity. As reported (20), destabilization of LSD1 occurs via
the ubiquitin-proteasome pathway by an E3 ubiquitin ligase, Jade-2
during neurogenesis, but the stabilization of LSD1 is unclear.
In the present study, we first discovered LSD1 as
the new substrate of USP7 from a molecular level. The evidence is
as follows. Firstly, we identified LSD1 as an associated protein by
immunoprecipitation with USP7 but no other DUB proteins, and GST
pull-down assays confirmed that LSD1 could interact with USP7
directly. Notably, overexpression of USP7 increased the protein
level of LSD1 but not the mRNA level. The mechanism involved is
that USP7 could protect ubiquitination and the proteasomal
degradation of LSD1. As reported, expression of USP7 in gliomas was
correlated with disease progression and patient survival time
(21), but the mechanism involved
is still unclear. In order to further understand the role of USP7
and LSD1 in glioblastoma progression, we investigated the
expression of USP7 and LSD1 in 150 cases of human glioma and normal
brain tissues and our data demonstrated that USP7 and LSD1 mRNA
expression was higher compared with the normal brain tissues with
an increasing trend from grade I to grade IV glioma according to
WHO classification. Furthermore, the expression of LSD1 was
positively correlated with USP7 expression in human glioma. These
results suggested that the stabilization of LSD1 by USP7 may
participate in glioma progression. We defined the function of USP7
and LSD1 in cell proliferation and invasion using GBM cell lines
A172 and T98G, as the ectopic expression of LSD1 could partly
induce the tendency by USP7 knockdown in the proliferation and
invasion of GBM cells. As reported, LSD1 mediated various
epigenetic gene expression regulating effects, such as p53
(22). In this study, we confirmed
that p53 is a key downstream transcription factor that mediates the
action of USP7 and LSD1. The aforementioned experiments further
confirmed that the regulation of LSD1 by USP7 plays an important
role in GBM and may function as a therapeutic target in GBM.
Acknowledgments
This study was supported by HuNan Provincial
Innovation Foundation for Postgraduate (No. CX2015B062).
References
|
1
|
Lan F, Nottke AC and Shi Y: Mechanisms
involved in the regulation of histone lysine demethylases. Curr
Opin Cell Biol. 20:316–325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prusevich P, Kalin JH, Ming SA, Basso M,
Givens J, Li X, Hu J, Taylor MS, Cieniewicz AM, Hsiao PY, et al: A
selective phenelzine analogue inhibitor of histone demethylase
LSD1. ACS Chem Biol. 9:1284–1293. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Amente S, Milazzo G, Sorrentino MC,
Ambrosio S, Di Palo G, Lania L, Perini G and Majello B:
Lysine-specific demethylase (LSD1/KDM1A) and MYCN cooperatively
repress tumor suppressor genes in neuroblastoma. Oncotarget.
6:14572–14583. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X
and Song Y: Over-expression of LSD1 promotes proliferation,
migration and invasion in non-small cell lung cancer. PLoS One.
7:e350652012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang Z, Li S, Song W, Li X, Li Q, Zhang
Z, Han Y, Zhang X, Miao S, Du R, et al: Lysine-specific demethylase
1 (LSD1/KDM1A) contributes to colorectal tumorigenesis via
activation of the Wnt/β-catenin pathway by down-regulating
Dickkopf-1 (DKK1) [corrected]. PLoS One. 8:e700772013. View Article : Google Scholar
|
|
6
|
Ketscher A, Jilg CA, Willmann D, Hummel B,
Imhof A, Rüsseler V, Hölz S, Metzger E, Müller JM and Schüle R:
LSD1 controls metastasis of androgen-independent prostate cancer
cells through PXN and LPAR6. Oncogenesis. 3:e1202014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu
W, Liang J, Sun L, Yang X, Shi L, et al: LSD1 is a subunit of the
NuRD complex and targets the metastasis programs in breast cancer.
Cell. 138:660–672. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Si W, Huang W, Zheng Y, Yang Y, Liu X,
Shan L, Zhou X, Wang Y, Su D, Gao J, et al: Dysfunction of the
reciprocal feedback loop between GATA3- and zEB2-nucleated
repression programs contributes to breast cancer metastasis. Cancer
Cell. 27:822–836. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Singh MM, Manton CA, Bhat KP, Tsai WW,
Aldape K, Barton MC and Chandra J: Inhibition of LSD1 sensitizes
glioblastoma cells to histone deacetylase inhibitors. Neuro Oncol.
13:894–903. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li M, Chen D, Shiloh A, Luo J, Nikolaev
AY, Qin J and Gu W: Deubiquitination of p53 by HAUSP is an
important pathway for p53 stabilization. Nature. 416:648–653. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brooks CL, Li M, Hu M, Shi Y and Gu W: The
p53-Mdm2-HAUSP complex is involved in p53 stabilization by HAUSP.
Oncogene. 26:7262–7266. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saridakis V, Sheng Y, Sarkari F, Holowaty
MN, Shire K, Nguyen T, Zhang RG, Liao J, Lee W, Edwards AM, et al:
Structure of the p53 binding domain of HAUSP/USP7 bound to
Epstein-Barr nuclear antigen 1 implications for EBV-mediated
immortalization. Mol Cell. 18:25–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song MS, Salmena L, Carracedo A, Egia A,
Lo-Coco F, Teruya-Feldstein J and Pandolfi PP: The
deubiquitinylation and localization of PTEN are regulated by a
HAUSP-PML network. Nature. 455:813–817. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maertens GN, El Messaoudi-Aubert S,
Elderkin S, Hiom K and Peters G: Ubiquitin-specific proteases 7 and
11 modulate Polycomb regulation of the INK4a tumour suppressor.
EMBO J. 29:2553–2565. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van der Horst A, de Vries-Smits AM,
Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM
and Burgering BM: FOXO4 transcriptional activity is regulated by
monoubiquitination and USP7/HAUSP. Nat Cell Biol. 8:1064–1073.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang Z, Wu Q, Guryanova OA, Cheng L, Shou
W, Rich JN and Bao S: Deubiquitylase HAUSP stabilizes REST and
promotes maintenance of neural progenitor cells. Nat Cell Biol.
13:142–152. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Erpolat OP, Akmansu M, Goksel F, Bora H,
Yaman E and Büyükberber S: Outcome of newly diagnosed glioblastoma
patients treated by radiotherapy plus concomitant and adjuvant
temozolomide: A long-term analysis. Tumori. 95:191–197.
2009.PubMed/NCBI
|
|
19
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al European Organisation for Research and Treatment of Cancer
Brain Tumor and Radiotherapy Groups; National Cancer Institute of
Canada Clinical Trials Group: Radiotherapy plus concomitant and
adjuvant temozolomide for glioblastoma. N Engl J Med. 352:987–996.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han X, Gui B, Xiong C, Zhao L, Liang J,
Sun L, Yang X, Yu W, Si W, Yan R, et al: Destabilizing LSD1 by
Jade-2 promotes neurogenesis: An antibraking system in neural
development. Mol Cell. 55:482–494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng C, Niu C, Yang Y, Wang Y and Lu M:
Expression of HAUSP in gliomas correlates with disease progression
and survival of patients. Oncol Rep. 29:1730–1736. 2013.PubMed/NCBI
|
|
22
|
Periz G, Lu J, Zhang T, Kankel MW,
Jablonski AM, Kalb R, McCampbell A and Wang J: Regulation of
protein quality control by UBE4B and LSD1 through p53-mediated
transcription. PLoS Biol. 13:e10021142015. View Article : Google Scholar : PubMed/NCBI
|