Introduction
The formation of metastases comprises multiple
sequential steps by which malignant cells disseminate from the
primary tumour, invade the surrounding stroma, breach the
vasculature, travel through the fluids, extravasate and colonise
distant organs (1). Colorectal
cancer (CRC) and breast cancer colonise distant organs through the
lymphatic vasculature. Whereas CRC frequents also blood vessels for
metastatic spreading, it is widely accepted that this route in not
common for breast cancer. A validated in vitro assay
recapitulates the preference of breast cancer cell spheroids to
breach the lymphatic endothelial cell (LEC) barrier but not the
blood endothelial cell (BEC) barrier (2). The quantitative assay was further
developed to study the malignant potential of CRC spheroids formed
by CCL227 cells, or by the 5-fluorouracil (5-FU)-resistant (5-FU is
a standard drug for CRC treatment) subclone CCL227-RH (3), to study breaching of the BEC barrier
and to define potential intervention strategies. Acquisition of
drug resistance is a complex process that profoundly alters gene
expression at the genetic and epigenetic level and impacts on cell
behaviour such as epithelial-to-mesenchymal transition (EMT) and
cell mobility. Reportedly, 5-FU-resistant CRC cells breach LEC
barriers significantly faster than naïve CRC cells (4). This is due to low levels of
microRNA200 (miR200) in those exosomes secreted by 5-FU-resistant
CCL227-RH cells (5). The
exosome-encapsulated miR200 family includes five members encoded by
two genes on chromosome 1 (miR200b, miR200a, miR429) and chromosome
12 (miR200c, miR141) (6) which
downregulate the expression of the ZEBs and are inducers of EMT
being a key process associated with the progression of CRC
(7,8). CRC-derived miR200 is taken up by
adjacent LECs, which suppresses ZEB transcription factors thereby
reducing plasticity and migration of LEC and slowing down the
formation of gates within the LEC barrier through which the tumour
can traverse (4). Therefore,
metastasis formation is not only an active process of the malignant
cell, but also an orchestrated and transient takeover of the
endothelial barrier.
Since CRC colonises distant organs through both,
lymphatics and blood vessels, we attempted to further validate the
new CRC model and tested how BECs respond to naïve and resistant
CRC spheroids and examined which of miR200 family members impose
the strongest effect on the ZEB expression in BECs.
Materials and methods
Reagents and antibodies
Sulforaphane was from Sigma-Aldrich Chemie GmbH
(Munich, Germany; R,S-sulforaphane, a racemic mixture of R and S
isomers; the R isomer is biologically active), mocetinostat was
from Chemietek (Indianapolis, IN, USA). Rabbit anti-zinc finger
E-box-binding homeobox 1 (ZEB1), and goat anti-SLUG were from Santa
Cruz Biotechnology, Inc. (Heidelberg, Germany). Rabbit anti-ZEB2
was from Merck Millipore (Darmstadt, Germany), mouse
anti-VE-cadherin was from Beckman Coulter (Fullerton, CA, USA),
rabbit anti-TWIST was from Abcam and rabbit anti-SNAI was from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Monoclonal mouse
anti-β-actin (clone AC-15) was from Sigma-Aldrich Chemie GmbH and
peroxidase-conjugated polyclonal swine anti-rabbit IgG (1:5,000),
peroxidase-conjugated polyclonal rabbit anti-mouse IgG (1:10,000),
peroxidase-conjugated polyclonal rabbit anti-goat IgG (1:10,000)
were from DakoCytomation (Glostrup, Denmark).
Cell culture
The CRC cell line CCL227 (lymph node metastasis) was
purchased from the American Type Culture Collection (Rockville, MD,
USA). From this cell line the highly 5-FU-resistant subclone
CCL227-RH (resistant against 125 µM 5-FU) was selected by
continuous addition of 5-FU over a period of time. Only when used
for experiments 5-FU was withdrawn from cell cultures several days
before. All cells were grown in RPMI-1640 medium supplemented with
10% heat-inactivated fetal calf serum, 25 mg/ml gentamycin (G418)
(all from Gibco, Karlsruhe, Germany) and maintained in humidified
atmosphere containing 5% CO2 at 37°C. The subclone
CCL227-RH was continuously grown in the presence of 5-FU.
Telomerase-immortalised human BECs were grown in EGM-2 MV
(EBM2-based medium CC-3156 and supplement CC-4147; Clonetics,
Allendale, NJ, USA) and G418. BECs were generated from human dermal
microvascular endothelial cells (C-12260) obtained from PromoCell
GmbH (Heidelberg, Germany) and telomerase-immortalized as described
earlier (9).
Spheroid formation
A total of 2×104 cells/ml (2,000
cells/spheroid) of the cell lines CCL227 or CCL227-RH was
transferred to RPMI medium containing 20% methylcellulose solution
(final concentration 0.3%). One hundred and fifty microliters of
this cell suspension was added to each well of a 96-well plate
(CELLSTAR 650185; Greiner Bio-One, Kremsmünster, Austria) and
centrifuged 15 min at 1,200 rpm to allow spheroid formation within
6 days at 37°C and 5% CO2. To generate more stable and
compact spheroids 1.7% Matrigel was added.
Circular chemorepellent-induced defect
(CCID) assay
In this assay the sizes of the cell-free areas
forming directly underneath the CRC cell spheroids in the
endothelial monolayer (CCIDs) were measured. BECs were seeded into
24-well plates and grown to ~90% confluence. Then, they were
incubated with a 1:5,000 dilution of cyto-tracker green (Molecular
Probes Life Technologies, Carlsbad, CA, USA) for 1 h in EGM2V
medium. Spheroids were washed and transferred to the
cyto-tracker-stained BEC monolayer. After 3 h of co-cultivation,
the size of CCID areas underneath CRC cell spheroids were
photographed using a fluorescence microscope Axiovert and
calculated with the AxioVison Rel. 4.8 program (both from Carl
Zeiss AG, Jena, Germany). The CCID areas underneath 10 or more
spheroids were measured.
Spheroid treatment with inhibitors
CRC cell spheroids were collected and washed twice
with fresh medium to remove methylcellulose. Then the spheroids
were pre-treated with different inhibitors for 30 min in 1 ml EGM2V
medium. All inhibitors were dissolved fresh in DMSO and diluted to
the indicated final concentrations. The DMSO concentration of 0.1%
was kept constant. Afterwards, 1 ml medium and spheroids were
transferred to confluent cyto-tracker-stained BEC monolayers and
incubated for 3 h.
Western blotting
BECs were seeded in T-25 flasks and grown until 80%
confluence. BECs were either transfected with the miR200 family
precursors (5 nM of miR200a, miR200b, miR200c, miR141, and miR429)
for 24 h or treated with drugs for 0.5, 1, 2, 4 h. Then, cells were
washed twice with cold PBS, placed on ice and lysed with the 2X SDS
lysis buffer and sonication on ice. The protein concentration was
determined by Bio-Rad assay and measured with a photometer at 260
nm. Equal protein amounts were mixed with 5X loading dye and lysis
buffer to a final volume of 20 µl, denatured at 95°C for 5
min and electrophoretically separated by 10% polyacrylamide SDS
gels, electrotransferred to PVDF membranes (GE Healthcare Life
Sciences, Little Chalfont, UK) as described earlier (10) and immunoblots were visualized using
ECL detection kit (Thermo Scientific, Portsmouth, IL, USA) and
Amersham Hyperfilm (GE Healthcare, Buckinghamshire, UK).
MicroRNA transfection
BECs were transfected using siPORT NeoFX (Ambion
Life Technologies, Carlsbad, CA, USA) at a confluence of ~80%. CRC
cell spheroids were cultivated for 6 days when they were
transfected. All microRNA precursors (miR200a, miR200b, miR200c,
miR141, miR429) and non-targeting control RNA were diluted (in 100
µl) to a final concentration of 5 nM in RPMI or EGM2V
without serum and antibiotics. A total of 10 µl siPORT
reagent was mixed with 90 µl of RPMI and mixed 1:1 with the
diluted microRNA solution. Thereafter, the solution was incubated
for 10 min at room temperature. The mixture was gently added to
either the cells or the spheroids in 2.3 ml RPMI medium and after
24 h the medium was changed to complete RPMI medium.
Isolation of exosomes
CRC cells were grown in a T-75 flask until ~80% of
confluence. Afterwards, cells were washed with PBS and kept in RPMI
without FCS and antibiotics for 24 h when the medium was changed to
RPMI with 10% exosome-free FCS for further 24 h to allow exosome
production. Thereafter, the supernatant was collected, centrifuged
at 3,000 × g for 15 min and transferred to a new 15-ml tube. The
exosomes in the supernatant were isolated using 2 ml ExoQuick-TC
(System Biosciences, Mountain View, CA, USA) per 5 ml supernatant
and pelleted at 1,500 g, 4°C for 30 min and washed twice.
Afterwards the pellet was resuspended in 10–100 µl DEPC
water. The exosomal protein concentration was measured with a
photometric protein assay (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Dose-response curves were analysed using Excel 2013
software and GraphPad Prism 6 software package (GraphPad, Software,
Inc., San Diego, CA, USA). The values were expressed as means ± SEM
and significance was calculated by Student's t-test and ANOVA
(statistical significance p<0.05).
Results
Comparison of the in vitro responses of
LECs and BECs to CRC clones
MCF-7 and MDA-MB231 breast cancer spheroids trigger
the retraction of adjacent LEC monolayers which causes the
formation of CCIDs (10,11). A similar centrifugal migration of
LECs and BECs was seen in the CRC model and time-course experiments
were performed to investigate the activity of CCL227
spheroid-triggered breaching of the beneath growing LEC and BEC
barriers. 5-FU treatment of CRC causes the development of drug
resistance and also the aggressiveness of tumours increases.
Therefore, it was investigated whether spheroids of the highly
5-FU-resistant CCL227-RH subclone exhibited an elevated potential
in breaching endothelial barriers as compared to naïve CCL227
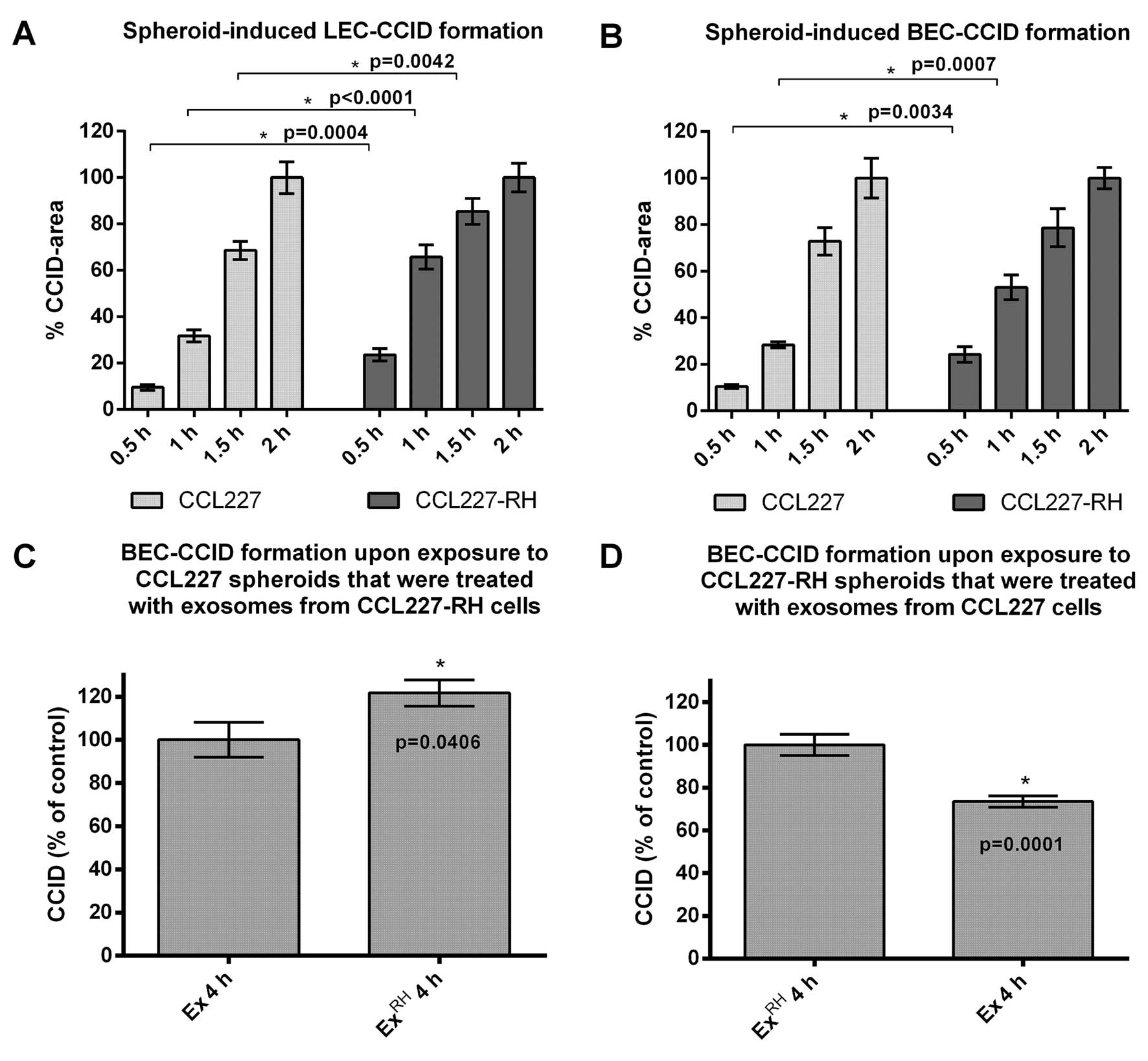
spheroids. In fact, CCL227-RH spheroids triggered CCID formation of
underneath growing LEC and BEC monolayers significantly faster than
naïve CCL227 spheroids (Fig. 1A and
B). In detail, CCL227-RH spheroids time-dependently induced
larger CCIDs than CCL227 spheroids in LECs (~15% after 0.5 h, ~35%
after 1 h and ~20% after 1.5 h) and in BECs (~15% after 0.5 h and
~25% after 1 h).
During the acquisition of 5-FU drug resistance
CCL227-RH cells have lost the expression of miR200 family members,
which are otherwise packed into exosomes and released from naïve
CCL227 cells (4). Hence, exosomes
derived from CCL227 cells are rich in miR200, whereas CCL227-RH
exosomes are depleted (5). miR200
family members downregulate ZEBs, induce E-cadherin, reduce
migration and limit EMT (12,13).
The downregulation of ZEB1 and upregulation of VE-cadherin in LECs
was shown to correlate with reduced CCID formation triggered by
MCF-7 spheroids (14). Therefore,
we assumed that miR200-enriched exosomes, which are shed by CRC
cells, act as potential modulators of BEC mobility and focussed on
their effects on CCID formation in BEC monolayers. When naïve
CCL227 spheroids were pre-incubated with exosomes derived from
resistant CCL227-RH cells an increase of CCID formation (~20%) was
observed after 4 h (Fig. 1C). We
think that this effect was due to a dilution of miR200-containing
exosomes of naïve CCL227 cells by miR200-depleted exosomes from
5-FU-resistant CCL227-RH cells. Reciprocally, the pre-incubation of
CCL227-RH spheroids with exosomes collected from CCL227 cells,
which are rich in miR200 (4,5),
reduced the CCID-forming potential of the CCL227-RH clone by 20%
(Fig. 1D) and this was most likely
due to a concentration effect of miR200-enriched exosomes sticking
to the spheroid surface.
The miR200 family inhibits ZEB
transcription factor expression and CCID formation in BECs
Members of the miR200 family comprising miR200a,
miR200b, miR200c, miR141 and miR429 regulate EMT (6). To examine which of these members
impose the strongest effect on the ZEB family of transcription
factors in BECs, 5 nM miR precursors were separately transfected
and the expression of ZEB1, ZEB2, SNAI, TWIST (SLUG was not
expressed) and VE-cadherin was analysed by western blotting
(Fig. 2A). The immunoblots
indicated a repression of ZEB1, ZEB2, SNAI and TWIST upon
transfection of all five miR200 family members. ZEB1 was repressed
by miR200a and miR200b, TWIST was inhibited upon transfection with
miR200c and miR141, whereas ZEB2 and SNAI were most effectively
inhibited by miR200c, miR141 and miR429. VE-cadherin expression was
almost unaffected in BECs and similar to that in LECs (4). Since CRC-derived miR200c, miR141 and
miR429 were the strongest suppressors of ZEB2 and SNAI in BECs, we
focussed on the stromal effects of these transcription factors and
how this influenced CCID formation in the CRC/BEC model. For this,
resistant CCL227-RH spheroids were transfected with the individual
precursor microRNAs and placed on BEC monolayers. miR200c, miR141
and miR429 inhibited CCID formation significantly (30, 20 and 20%,
respectively), whereas miR200a and miR200b induced CCID formation
(Fig. 2B). This suggested that
inhibition of ZEB1 by miR200a and miR200b did not play a role in
stabilising the BEC barriers, whereas miR200c, miR141 and miR429,
which inhibited ZEB2 and SNAI in BECs, attenuated their
disintegration.
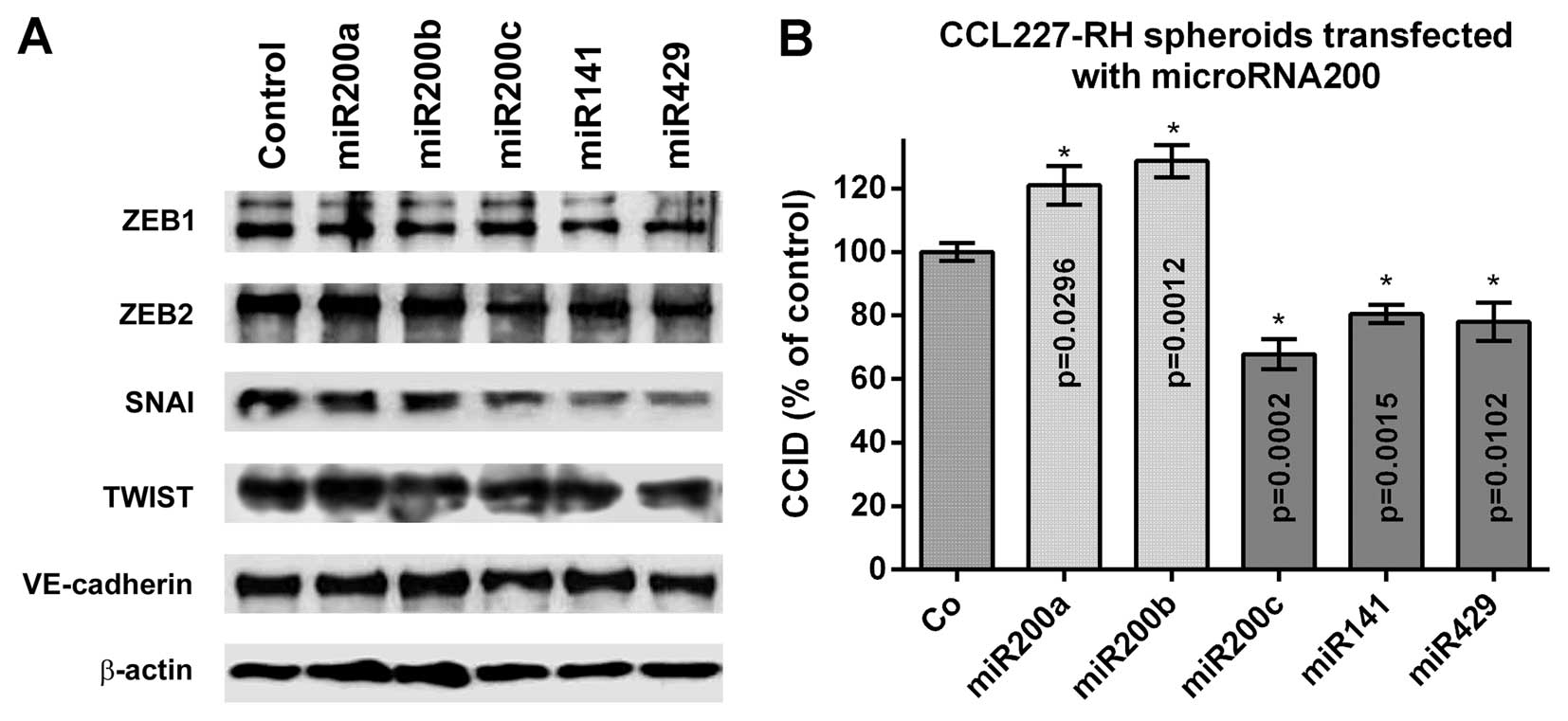 | Figure 2Effect of miR200 family members on the
expression of EMT markers and CCID formation. (A) Expression of the
EMT-related transcription factors ZEB1, ZEB2, SNAI, TWIST and of
VE-cadherin in BECs, which were transfected with 5 nM miR200 family
precursors or non-targeting control, was analysed by western
blotting. (B) 5-FU-resistant CCL227-RH spheroids were separately
transfected with 5 nM miR200a, miR200b, miR200c, miR141 and miR429
precursors or non-targeting control RNA (Co) and placed on the BEC
monolayer and CCID formation was measured; (n=3). Error bars
indicate means ± SEM and asterisks indicate significance
(p<0.05; Student's t-test). miR200, microRNA200; EMT,
epithelial-to-mesenchymal transition; CCID, circular
chemorepellent-induced defect; ZEB1, zinc finger E-box-binding
homeobox 1; BECs, blood endothelial cells; 5-FU,
5-fluorouracil. |
Drug-dependent inhibition of CRC
spheroid-triggered disintegration of BEC barriers
It was reported that loss of histone acetylation
during CRC development correlates with advanced tumour stage and
tumour invasion, because a reduction in global acetylation of
histone H4 was observed in 80% of colon carcinomas and 39% of
adenomas (15). Hence, HDAC
inhibitors are receiving increasing interest as chemopreventive and
chemotherapeutic agents. With the goal to re-express miR200 family
members and to reduce malignancy the CRC/BEC model was tested with
the class I HDAC inhibitor mocetinostat and with the HDAC inhibitor
sulforaphane, which is a natural secondary metabolite present
mostly in cruciferous vegetables and in particularly high quantity
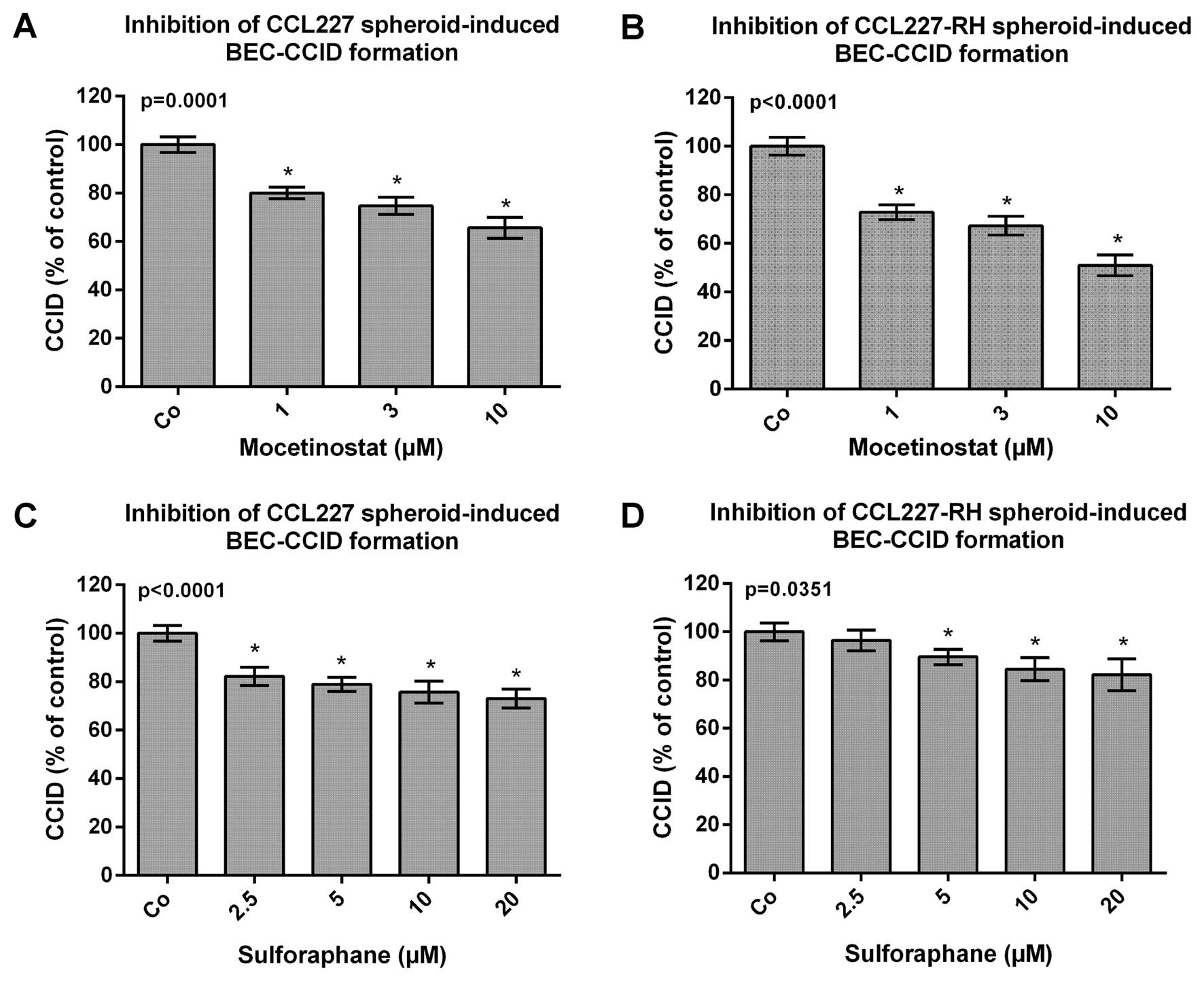
in broccoli sprouts (15,16). Thus, CCL227 and CCL227-RH spheroids
were pre-treated with mocetinostat and sulforaphane for 20 min and
then placed on BEC monolayers to analyse CCID formation. The
migration of BECs, induced by CCL227 and CCL227-RH spheroids, was
reduced by mocetinostat and sulforaphane in a dose-dependent
manner. Both compounds were non-toxic during the treatment time at
the used concentrations (data not shown). Mocetinostat inhibited
CCL227-RH-induced CCIDs more efficiently (30, 35 and 40%) than
CCIDs induced by CCL227 spheroids (20, 25 and 30%) (Fig. 3A and B). In contrast, sulforaphane,
which was less active than mocetinostat, inhibited
CCL227-RH-induced CCID formation by only 20% and CCL227-induced
CCIDs by 20–25% (Fig. 3C and
D).
Discussion
The present study investigated the response of BECs
upon contact with naïve CRC spheroids or with spheroids consisting
of high 5-FU-resistant CRC cells and confirmed earlier data showing
that the mobility of LECs was increased in the neighbourhood of
aggressive 5-FU-resistant CRC-RH cells as compared to less
aggressive naïve CRC cells (4).
Furthermore, the CRC/LEC and CRC/BEC in vitro models
recapitulated the pathologic situation in which CRC spreads through
lymphatics as well as through blood vessels. In contrast, it was
demonstrated that LEC barriers were about five times more sensitive
to breast cancer spheroid-induced CCID formation than BEC barriers
resembling the metastatic route of breast cancer cells through
lymphatics rather that blood vessels (2). Altogether, these findings underscore
the validity of the three-dimensional (3D) CCID assay as a tool to
study tumour breaching through the endothelial vasculature in
vitro.
Recently, it was reported that CCL227 cells secrete
exosomes containing miR200 family members whereas the exosomes of
5-FU-resistant CCL227-RH cells are virtually free of miR200, and
that miR200c inhibits CCID formation in the CRC/LEC model (4). Here we demonstrate that miR200c,
miR141 and miR429 (but not miR200a and miR200b) inhibit CCID
formation also in the CRC/BEC model. In BECs miR200c, miR141 and
miR429 inhibited ZEB2 and SNAI most effectively. SNAI plays a key
role in invasion and metastases of CRC, which is driven by the
TGF-β1/SMAD signalling pathway (17). Furthermore, SNAI specifically
represses vitamin D receptor, which itself mediates antitumour
action and is lost in progressive CRC (18). Upon SNAI-induced EMT the synthesis
of ZEB2 is upregulated (19) and
miR132 (which does not belong to the miR200 family) inhibits CRC
invasion and EMT by targeting ZEB2 (20). Hence, SNAI and ZEB2 significantly
contribute to EMT, endothelial breaching and metastasis of CRC.
Cells in the invasive front of colorectal tumours that have lost
expression of the miR200 family members are more likely to
intravasate into the blood or lymphatic system (6). Hence, malignant progression was
attenuated through horizontal transfer of miR200c, miR141 and
miR429 to an alien cell type. The downregulation of ZEBs upon
transfection of miR200 family members into BECs was almost similar
compared to LECs (4). As an
exception, miR200a and miR200b did not influence ZEB2 expression
and miR200c inhibited SNAI expression only weakly in LECs, whereas
ZEB2 and SNAI were inhibited by these microRNAs in BECs. We cannot
explain the inducing effect of miR200a and miR200b on CCID
formation but it seems that gene regulation by these microRNAs is
more complex than expected. Unlike the negative regulation of
E-cadherin by ZEBs in epithelial cells, VE-cadherin was not
suppressed by ZEBs in endothelial cells, which was supported by the
fact that knockdown of ZEB1 did not influence the expression of
VE-cadherin in LECs (14).
Therefore, other mechanisms must have been involved in the
regulation of VE-cadherin in BECs.
The class I HDAC inhibitor mocetinostat was shown to
lead to a re-expression of miR200 family members which
re-establishes sensitivity to gemcitabine-induced apoptosis of
otherwise treatment-resistant pancreatic cancer cells (21). Acetylation/deacetylation events go
hand in hand also with DNA methylations/demethylations, which was
recently observed for the miR141 and miR200c loci in vitro
and in vivo (22). Currently
mocetinostat is studied in 17 clinical trials (https://clinicaltrials.gov/ct2/results?term=mocetinostat&Search=Search).
Sulforaphane is also reported to cause a global
increase in histone H3 and H4 acetylation in HT-29 CRC cells
similar to mocetinostat in pancreatic cancer cells (23). This led to the assumption that
mocetinostat and sulforaphane might upregulate miR200 in BECs and
influence CCID formation. The short treatment time with
mocetinostat (~3.5 h) seemed to exclude an epigenetic rearrangement
of the chromatin as being causal for miR200 re-expression and for
reduced aggressiveness. Nevertheless, it was shown that a single
oral dose of sulforaphane was sufficient to inhibit HDAC activity
and rearrange chromatin in mouse colon causing an increase in
acetylated histones after 6 h (16)
and the activation of the promoter regions of p21 and
Bax genes implicates epigenetic alterations within such a
short treatment period (15).
Hence, also short treatment time allows the re-expression of
silenced genes. However, neither mocetinostat nor sulforaphane
caused a significant change in the expression of miR200a, miR200b,
miR200c, miR141 and miR429. Therefore, the inhibition of CCIDs upon
treatment with mocetinostat and sulforaphane was independent of
their anticipated effect on miR200 expression. Sulforaphane was
shown in Caco-2 cells to exert also another pharmacologic activity,
i.e., the induction of UGT1A1 and GSTA1 transcription
(detoxification enzymes) (24). As
the disintegration of BEC barriers in vitro was attenuated
by sulforaphane, which can be consumed by eating broccoli sprouts
in quantities that change the expression in human PBMCs in
vivo (16) and suppresses
tumorigenesis in rodents (15), and
by mocetinostat currently tested in clinical trials, both compounds
may provide a tool to support endothelial integrity. Whether miR200
expression has diagnostic/therapeutic impact regarding the
development of drug resistance of CRC remains to be
established.
Acknowledgments
We wish to thank Toni Jäger for preparing the
figures. C.H.N. was supported by technology grant (TSA Doktorat)
financed by the Austrian Federal Ministry of Science and Research
(BMFW) in frame of Asea Uninet. The study was supported by a grant
of the Herzfelder'sche Family Foundation to G.K.
References
|
1
|
de Krijger I, Mekenkamp LJM, Punt CJA and
Nagtegaal ID: MicroRNAs in colorectal cancer metastasis. J Pathol.
224:438–447. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kerjaschki D, Bago-Horvath Z, Rudas M,
Sexl V, Schneckenleithner C, Wolbank S, Bartel G, Krieger S, Kalt
R, Hantusch B, et al: Lipoxygenase mediates invasion of
intrametastatic lymphatic vessels and propagates lymph node
metastasis of human mammary carcinoma xenografts in mouse. J Clin
Invest. 121:2000–2012. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tentes IK, Schmidt WM, Krupitza G, Steger
GG, Mikulits W, Kortsaris A and Mader RM: Long-term persistence of
acquired resistance to 5-fluorouracil in the colon cancer cell line
SW620. Exp Cell Res. 316:3172–3181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Senfter D, Holzner S, Kalipciyan M,
Staribacher A, Walzl A, Huttary N, Krieger S, Brenner S, Jäger W,
Krupitza G, et al: Loss of miR-200 family in 5-fluorouracil
resistant colon cancer drives lymphendothelial invasiveness in
vitro. Hum Mol Genet. 24:3689–3698. 2015.PubMed/NCBI
|
|
5
|
Mader RM, Wieser M, Berger W, Kalipciyan
M, Hackl M, Steger GG and Grillari J: Relevance of microRNA
modulation in chemoresistant colon cancer in vitro. Int J Clin
Pharmacol Ther. 49:67–68. 2011.
|
|
6
|
Paterson EL, Kazenwadel J, Bert AG,
Khew-Goodall Y, Ruszkiewicz A and Goodall GJ: Down-regulation of
the miRNA-200 family at the invasive front of colorectal cancers
with degraded basement membrane indicates EMT is involved in cancer
progression. Neoplasia. 15:180–191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Findlay VJ, Wang C, Watson DK and Camp ER:
Epithelial-to-mesenchymal transition and the cancer stem cell
phenotype: Insights from cancer biology with therapeutic
implications for colorectal cancer. Cancer Gene Ther. 21:181–187.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sánchez-Martínez R, Cruz-Gil S, Gómez de
Cedrón M, Álvarez-Fernández M, Vargas T, Molina S, García B,
Herranz J, Moreno-Rubio J, Reglero G, et al: A link between lipid
metabolism and epithelial-mesenchymal transition provides a target
for colon cancer therapy. Oncotarget. 6:38719–38736.
2015.PubMed/NCBI
|
|
9
|
Schoppmann SF, Soleiman A, Kalt R, Okubo
Y, Benisch C, Nagavarapu U, Herron GS and Geleff S:
Telomerase-immortalized lymphatic and blood vessel endothelial
cells are functionally stable and retain their lineage specificity.
Microcirculation. 11:261–269. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nguyen CH, Senfter D, Basilio J, Holzner
S, Stadler S, Krieger S, Huttary N, Milovanovic D, Viola K,
Simonitsch-Klupp I, et al: NF-κB contributes to MMP1 expression in
breast cancer spheroids causing paracrine PAR1 activation and
disintegrations in the lymph endothelial barrier in vitro.
Oncotarget. 6:39262–39275. 2015.PubMed/NCBI
|
|
11
|
Madlener S, Saiko P, Vonach C, Viola K,
Huttary N, Stark N, Popescu R, Gridling M, Vo NT, Herbacek I, et
al: Multifactorial anticancer effects of digalloyl-resveratrol
encompass apoptosis, cell-cycle arrest, and inhibition of
lymphendothelial gap formation in vitro. Br J Cancer.
102:1361–1370. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Loboda A, Nebozhyn MV, Watters JW, Buser
CA, Shaw PM, Huang PS, Van't Veer L, Tollenaar RA, Jackson DB,
Agrawal D, et al: EMT is the dominant program in human colon
cancer. BMC Med Genomics. 4:92011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yuan D, Xia H, Zhang Y, Chen L, Leng W,
Chen T, Chen Q, Tang Q, Mo X, Liu M, et al: p-Akt/miR 200 signaling
regulates epithelial-mesenchymal transition, migration and invasion
in circulating gastric tumor cells. Int J Oncol. 45:2430–2438.
2014.PubMed/NCBI
|
|
14
|
Vonach C, Viola K, Giessrigl B, Huttary N,
Raab I, Kalt R, Krieger S, Vo TP, Madlener S, Bauer S, et al: NF-κB
mediates the 12(S)-HETE-induced endothelial to mesenchymal
transition of lymphendothelial cells during the intravasation of
breast carcinoma cells. Br J Cancer. 105:263–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Myzak MC, Dashwood WM, Orner GA, Ho E and
Dashwood RH: Sulforaphane inhibits histone deacetylase in vivo and
suppresses tumorigenesis in Apc-minus mice. FASEB J. 20:506–508.
2006.PubMed/NCBI
|
|
16
|
Myzak MC, Tong P, Dashwood WM, Dashwood RH
and Ho E: Sulforaphane retards the growth of human PC-3 xenografts
and inhibits HDAC activity in human subjects. Exp Biol Med
(Maywood). 232:227–234. 2007.
|
|
17
|
Ji Q, Liu X, Han Z, Zhou L, Sui H, Yan L,
Jiang H, Ren J, Cai J and Li Q: Resveratrol suppresses
epithelial-to-mesenchymal transition in colorectal cancer through
TGF-β1/Smads signaling pathway mediated Snail/E-cadherin
expression. BMC Cancer. 15:972015. View Article : Google Scholar
|
|
18
|
Larriba MJ, Martín-Villar E, García JM,
Pereira F, Peña C, de Herreros AG, Bonilla F and Muñoz A: Snail2
cooperates with Snail1 in the repression of vitamin D receptor in
colon cancer. Carcinogenesis. 30:1459–1468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Beltran M, Puig I, Peña C, García JM,
Alvarez AB, Peña R, Bonilla F and de Herreros AG: A natural
antisense transcript regulates Zeb2/Sip1 gene expression during
Snail1-induced epithelial-mesenchymal transition. Genes Dev.
22:756–769. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zheng YB, Luo HP, Shi Q, Hao ZN, Ding Y,
Wang QS, Li SB, Xiao GC and Tong SL: miR-132 inhibits colorectal
cancer invasion and metastasis via directly targeting ZEB2. World J
Gastroenterol. 20:6515–6522. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meidhof S, Brabletz S, Lehmann W, Preca
BT, Mock K, Ruh M, Schüler J, Berthold M, Weber A, Burk U, et al:
ZEB1-associated drug resistance in cancer cells is reversed by the
class I HDAC inhibitor mocetinostat. EMBO Mol Med. 7:831–847. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Díaz-Martín J, Díaz-López A, Moreno-Bueno
G, Castilla MÁ, Rosa-Rosa JM, Cano A and Palacios J: A core
microRNA signature associated with inducers of the
epithelial-to-mesenchymal transition. J Pathol. 232:319–329. 2014.
View Article : Google Scholar
|
|
23
|
Dashwood RH and Ho E: Dietary histone
deacetylase inhibitors: From cells to mice to man. Semin Cancer
Biol. 17:363–369. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Svehlíková V, Wang S, Jakubíková J,
Williamson G, Mithen R and Bao Y: Interactions between sulforaphane
and apigenin in the induction of UGT1A1 and GSTA1 in CaCo-2 cells.
Carcinogenesis. 25:1629–1637. 2004. View Article : Google Scholar : PubMed/NCBI
|