Introduction
Non-Hodgkin's lymphomas (NHLs), derived from B- or
T-cell, constitute a heterogeneous group of malignant
lymphoproliferative disorders (1,2), which
accounts for approximately 90% of all lymphomas (3). The incidence of NHLs has increased in
recent years and is still associated with significant mortality.
Despite intensive efforts in developing new therapies, the
emergence of clinical drug resistance remains a barrier to
successful treatment of lymphoma (4,5).
Recently, evidence has accumulated showing that lymphoma tumor
microenvironment provides sanctuary for lymphoma cells through
interaction which occurs between the lymphoma cell and its
microenvironment (lymph node stroma) (6). Adhesion of lymphoma cells to
microenvironment components is shown to be a critical determinant
for mediating tumor resistance to cytotoxic therapy (cell adhesion
mediated drug resistance; CAM-DR) (7). CAM-DR is one of the most important
mechanisms in tumor cell microenvironment protection. However, it
remains unclear how the lymphoma microenvironment influences
lymphoma cell survival and responses to therapy, as well as the
molecular mechanisms involved.
The PH domain-containing casein kinase 2 interacting
protein-1 (CKIP-1; also known as PLEKHO1) is involved in regulating
many processes such as cell proliferation, differentiation and
apoptosis (8). CKIP-1 also plays an
important role in many cancers, such as colon, breast cancer and
human osteosarcoma (9). CKIP-1 has
been reported to regulate numerous important proteins controlling
cell survival, such as CK2, Akt and Smurf1. It has been generally
demonstrated that the activities of CK2, Akt and Smurf1 are
elevated in many human cancers (10–12).
In addition, CKIP-1 negatively regulates several important
pathways, such as TGF-β/BMP signaling, and PI3K/Akt signaling,
indicating that CKIP-1 acts as an crucial master in tumorigenicity
control (11). A previous study
reported that CKIP-1 enhances sensitivity to chemotherapy drugs by
targeting the Akt PH domain and suppressing Akt kinase activity in
human cancers (10). It has been
reported that knockdown of CKIP-1 in colon cancer cells shortened
the G1/S phase and reduced cell proliferation due to decreased Akt
kinase activity (13). These
observations together revealed that CKIP-1 might function as a
potential tumor suppressor. However, the functional role of CKIP-1
in non-Hodgkins lymphoma (NHL) has never been elucidated.
Considering the pivotal role CKIP-1 played in tumorigenicity
control and the fact that CKIP-1 can regulate Akt activity, it
would be intriguing to verify whether CKIP-1 plays a key role in
CAM-DR in NHL.
In the present study, we aimed to investigate the
expression and function of CKIP-1 in NHL and CAM-DR phenotype. This
study demonstrated for the first time that CKIP-1 is a novel
prognostic indicator in NHL. Furthermore, we also demonstrated that
CKIP-1 plays a critical role in CAM-DR via interacting with Akt and
regulating Akt kinase activity in NHL. Taken together, this study
may provide a novel perspective for a better understanding of the
role for CKIP-1in NHL and CAM-DR.
Materials and methods
Cell culture
Burkitt lymphoma cell-line Raji and diffuse large
B-cell lymphoma (DLBCL) cell lines OCI-Ly8 were obtained from Fudan
University (Shanghai, China). Bone marrow stromal cell line HS-5
was obtained from Cell Library, China Academy of Science. The human
lymphoma cell lines were grown in suspension in RPMI-1640
(Sigma-Aldrich, Rehovot, Israel) and the HS-5 was grown in F12
(Sigma-Aldrich), supplemented with 10% fetal bovine serum (FBS;
Gibco-BRL, Grand Island, NY, USA) at 37°C and 5%
CO2.
Cell co-culture
Firstly, dishes were coated with 40 lg/ml human FN
(Sigma-Aldrich) or HS-5 cells at 37°C. Secondly, lymphoma cells
(105 cells/ml) were allowed to adhere to preincubated FN
or HS-5 for 24 h. Finally, adherent cells were removed for
subsequent experiments.
Western blot analysis and
antibodies
The following monoclonal antibodies were purchased:
CKIP-1 (Cell Signaling Technology, Beverly, MA, USA); AKT (Cell
Signaling Technology), Gsk-3β (Cell Signaling Technology),
anti-p-Akt (Ser473) and anti-pAkt (Thr308) (Cell Signaling
Technology) and pGsk-3β (Cell Signaling Technology); anti-β-actin
(Sigma-Aldrich). Protein was run on a 10% PAGE and transferred to
polyvinylidine difluoride filter (PVDF) membranes. The membranes
were blocked with phosphate-buffered saline (PBS) containing 5%
skim milk and 0.1% Tween-20 and then incubated with primary
antibody overnight at 4°C. After washing with PBS containing 0.1%
Tween-20 three times, each for 5 min, filters were incubated with
horseradish peroxidase-conjugated secondary antibody for 2 h at
room temperature. Values were responsible for at least three
independent experiments.
Transient transfection
Full-length CKIP-1 cDNA were amplified using PCR and
subcloned into pCMV-HA and pcDNA3.1 constructs. Control-siRNA and
CKIP-1-siRNA were purchased from Shanghai GenePharma, Co., Ltd.
(Shanghai, China). Transfection was performed using Lipofectamine
2000 transfection reagent (Invitrogen) according to the
manufacturers instructions. Then, siRNA-resistant CKIP-1 construct
(designated myc-rCKIP-1) was generated by introducing silent
mutations in the targeting regions for CKIP-1-siRNA. NHL cells were
plated in FBS free RPMI-1640 medium without antibiotics and cells
were transfected at 70–90% confluence. Transfection was carried out
according to the manufacturers instructions. Transfected cells were
used for subsequent experiments 48 h after transfection.
RNA isolation and reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA of Raji cells and OCI-Ly8 cells were
extracted by a TRIzol extraction kit according to the manufacturers
protocol. The total RNA was reverse-transcribed by the ThermoScript
RT-PCR system (Invitrogen, Carlsbad, CA, USA). cDNA was amplified
using the following primers: CKIP-1 F, 5-AATTCTGCGGGAAAGGGATTT-3
and R, 5-AACACCTCCTGACTGTTTTCTC-3; c-Myc F,
5-AATCCTGTACCTCGTCCGAT-3 and R, 5-TCTTCTC CACAGACACCACA-3; cyclin
D1 F, 5-TGCTACCGCACA ACGCA-3 and R, 5-TCAATCTGTTCCTGGCAGGC-3;
cyclin E F, 5-CTCCAGGAAGAGGAAGGCAA-3 and R,
5-TCGATTTTGGCCATTTCTTCA-3; GAPDH F, 5-AGG TCGGTGTGAACGGATTTG-3 and
R, 5-TGTAGACCATG TAGTTGAGGTCA-3. After amplification, the products
were separated on an agarose (1.5%) gel and visualized under UV
light.
Cell viability assay
The cells were seeded on a 96-well plate (Corning
Incorporated, Corning, NY, USA) with a density of
1×106/well in volumes of 100 µl and grew 12 h with or
without the addition of doxorubicin (Sigma-Aldrich). Then, cell
proliferation was measured using Cell Counting kit-8 (CCK-8;
Dojindo Laboratories, Kumamoto, Japan). The absorbance was read by
a fluorometer (CytoFluor; Applied Biosystems, Foster City, CA, USA)
at 450 nm with a reference wavelength of 630 nm.
Apoptosis measurements
Drug-induced apoptosis following exposure to
doxorubicin (Sigma-Aldrich) was detected in NHL cells. In short,
following 48 h of chemotherapy agent exposure, apoptotic cells were
detected using Annexin V-FLUOS Staining kit (Roche Diagnostics,
Indianapolis, IN, USA) according to the manufacturers protocol.
Cells were washed three times and were resuspended in 100 l of
Annexin V-FLUOS labeling solution at a concentration of
1×106 cells/ml incubate in the dark for 15 min. To
analyse apoptosis, flow cytometry (BD FACSAriaII) was performed to
analyze the labeled cells.
Immunoprecipitation
Immunoprecipitation was performed as previously
described (14). In short, cell
lysates was incubated with anti-CKIP-1 antibody or control IgG
(Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight.
Protein A/G (Sigma-Aldrich) was then added for 2 h at 4°C with
rocking. The precipitates were washed three times with
homogenization buffer and boiled for 10 min with SDS sample buffer
followed by western blot analysis.
Soft agar colony assays
In short, cells were resuspended at 1×103
cells in 0.5 ml of 0.35% agar solution (Sigma-Aldrich, St. Louis,
MO, USA) containing RPMI-1640 cell culture medium and layered on
top of a 0.8% agar layer in 24-well plates. Plates were then
maintained for 14 days at 37°C with 5% CO2. Cell
colonies were stained with 0.5% crystal violet and visualized by
microscopy.
Statistical analysis
All experiments were repeated at least three times.
All data are reported as mean ± SEM. The calculations were analyzed
using the Statistical Package for Social Science SPSS 19.0 software
(SPSS, Inc., Chicago, IL, USA). Statistical significance was set at
P<0.05.
Results
CKIP-1 expression levels were
deregulated in proliferating NHL
It has been reported that CKIP-1 is deregulated in
multiple malignant tumor cell lines and tissues, such as breast
(15), colon cancer (11) and human osteosarcoma (16). However, the expression of CKIP-1 in
NHL is still unknown. In the present study, we first investigated
the alterations of CKIP-1 during NHL progression. Raji and OCI-Ly8
cells were cultured in serum-free medium for 72 h, and released
with RPMI-1640 medium containing 10% FBS for the indicated period
of time. Flow cytometric analysis demonstrated that cells were
arrested in the G1 phase due to serum deprivation, and the cells
were released from the G1 phase and entered the S phase following
serum re-feeding (Fig. 1A and B).
Next, we analyzed the expression of CKIP-1 in proliferating NHL
cells. As expected, CKIP-1 levels were decreased gradually after
serum stimulation, which was in accordance with upregulation of
PCNA in both Raji and OCI-Ly8 cells (Fig. 1C-F). These data indicated that
CKIP-1 might promote proliferation of NHL cells.
Knockdown of CKIP-1 promotes cell
proliferation
The repressed CKIP-1 expression in the progression
of NHL inspired us to investigate whether CKIP-1 functions as a
candidate tumor suppressor. Thus, we asked whether CKIP-1 could
also have a role in regulation of NHL cell proliferation. To
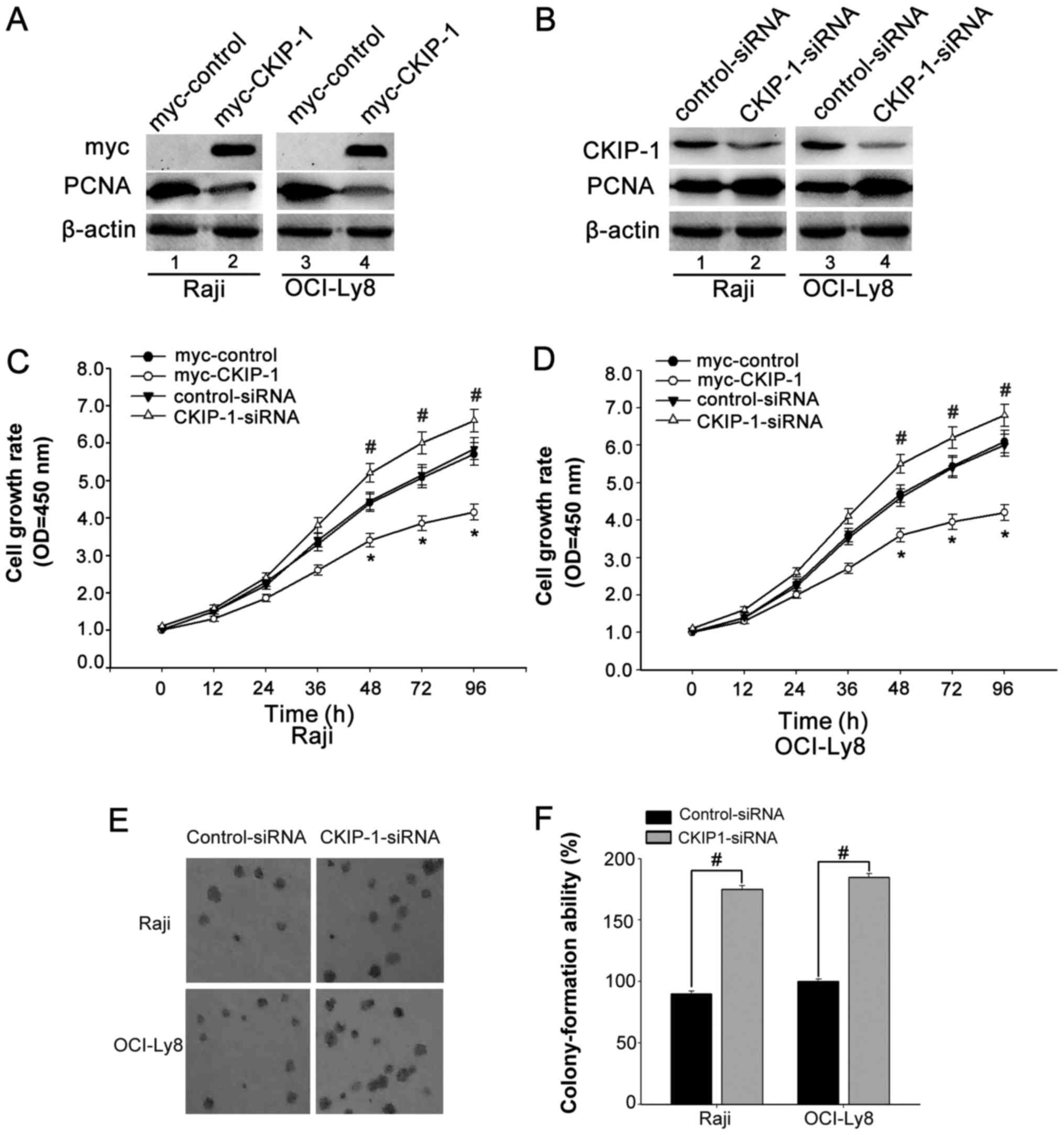
confirm our assumption, OCI-Ly8 and Raji cells were transfected
with CKIP-1-siRNA or myc-tagged CKIP-1 (myc-CKIP-1) or their
respective control. The interference efficiency was confirmed by
western blot analyses (Fig. 2A and
B). CCK-8 assays were used to investigate the effect of CKIP-1
on OCI-ly8 and Raji cell growth. We observed that cells were
transfected with myc-CKIP-1 showed reduced cell growth. On the
contrary, cells were transfected with CKIP-1-siRNA exhibited
increased cell growth (Fig. 2C and
D). In addition, colony formation assay showed that knockdown
of CKIP-1 resulted in a significant promotion of cell growth
(Fig. 2E and F). All the data
suggested that CKIP-1 might play a role in regulating cell
proliferation.
Knockdown of CKIP-1 promotes cell
cycle progression
To further confirm the effect of CKIP-1 on cell
cycle regulation, flow cytometric analysis was conducted and showed
that CKIP-1 generate a decrease in the percentage of cells in the
G1 phase when OCI-Ly8 and Raji cells were transfected with
CKIP-1-siRNA, but more cells were found in the G1 phase in the
presence of myc-CKIP-1 (Fig. 3A and
B). To further understand the effect of CKIP-1 on cell cycle
progression, we measured the cell cycle-regulatory protein
expression levels in NHL. RT-PCR revealed that cells were
transfected with CKIP-1-siRNA expressed more c-Myc, cyclin D1 and
cyclin E compared with control-siRNA transfected ones (Fig. 3C). Moreover, western blot analysis
showed that knockdown of CKIP-1 resulted in a significant increase
of c-Myc, cyclin D1, cyclin E and PCNA expression, whereas
overexpression of CKIP-1 resulted in a significant decrease of
their expression (Fig. 3D). All the
data indicate that knockdown of CKIP-1 in NHL cells promotes cell
cycle progression.
CKIP-1 reverses CAM-DR in NHL
A previous study reported that CKIP-1 is correlated
to chemotherapy in human cancer cells (8,10,11).
To determine whether CKIP-1 was associated with CAM-DR in NHL, cell
adhesion assays were first performed to assess the expression of
CKIP-1. Western blot analysis and RT-PCR analysis showed that
CKIP-1 expression was less obvious when cells adhered to FN or HS-5
cells compared with those cultured in suspension (Fig. 4A and B). To confirm whether CKIP-1
had an effect on CAM-DR in NHL, we prepared myc-tagged plasmid
expressing CKIP-1, which is resistant to silencing by CKIP-1-siRNA
(designated myc-rCKIP-1). The transfection efficiencies were
confirmed by western blot analysis (Fig. 4C). Then, Raji and OCI-Ly8 cells were
treated with 1 µM doxorubicin (Doxo). CCK-8 assays showed that
adhesion to FN or HS-5 cells significantly protected OCI-Ly8 and
Raji cells from chemotherapeutics compared to those cultured in
suspension. In addition, we found that this protective effect was
increased in cells transfected with CKIP-1-siRNA. Notably,
presentation of the myc-rCKIP-1 in CKIP-1-depleted cells abrogated
the protective effect (Fig. 4D).
Subsequently, flow cytometric analysis also showed that knockdown
of CKIP-1 in Raji and OCI-Ly8 cells adhered to FN or HS-5 cells
significantly decreased cell apoptosis. On the contrary, the
presentation of the myc-rCKIP-1 in CKIP-1-depleted cells increased
cell apoptosis (Fig. 4E). These
data suggested that overexpression of CKIP-1 could reverse CAM-DR
in NHL.
 | Figure 4.CKIP-1 reverses CAM-DR in NHL. (A and
B) Raji and OCI-Ly8 cells were cultured in suspension or adhered to
FN or HS-5 cells. Cells were analyzed for CKIP-1 and β-actin
expression by western blot analysis. Total RNAs were analyzed by
RT-PCR using CKIP-1-specific or GAPDH-specific primers. (C) Raji
and OCI-Ly8 cells were transfected with control-siRNA, CKIP-1-siRNA
or co-transfected with CKIP-1-siRNA and myc-tagged siRNA-resistant
CKIP-1 (designated myc-rCKIP-1-1). Raji and OCI-Ly8 cells were
analyzed for CKIP-1 and β-actin expression by western blot
analysis. (D) Raji and OCI-Ly8 cells were transfected with
control-siRNA, CKIP-1-siRNA or co-transfected with CKIP-1-siRNA and
GFP-rCKIP-1, then cells were adhered to FN, HS-5 cells or cultured
in suspension along with addition of 1 µM doxorubicin (Doxo). Then,
CCK-8 assays were performed to determine the cell viability. Data
are presented as mean ± SEM of three independent measurements
(*,#P<0.05). (E) Raji and OCI-Ly8 cells were
transfected with control-siRNA, CKIP-1-siRNA or co-transfected with
CKIP-1-siRNA and GFP-rCKIP-1, Raji and OCI-Ly8 cells were then
adhered to FN, HS-5 cells or cultured in suspension together with
addition of 1 µM Doxo. Flow cytometric analysis was used to
evaluate Doxo-induced cell apoptosis. Data are presented as mean ±
SEM of three independent measurements
(*,#P<0.05). |
CKIP-1 negatively regulates the
PI3K/Akt pathway by interacting with Akt
A previous study demonstrated that CKIP-1 interacts
with Akt, inhibits Akt phosphorylation and negatively regulates the
PI3K/Akt pathway in breast cancer cells (15). Thus, we wondered whether CKIP-1
overcame CAM-DR phenotype by regulating PI3K/Akt pathway. Firstly,
we sought to determine whether CKIP-1 could interact with Akt in
NHL. The immunoprecipitation experiment showed that Akt could
interact with CKIP-1 in OCI-Ly8 (Fig.
5A). Subsequently, Raji and OCI-Ly8 cells were transfected
myc-CKIP-1 in different doses. Western blot analysis revealed that
cells transfected with myc-CKIP-1 markedly reduced the
phosphorylated Akt levels at Thr308 and Ser473 in a dose-dependent
fashion. Consistent with the decrease in phosphorylated Akt levels,
cells expressing myc-CKIP-1 decreased the phosphorylation of GSK-3β
in a dose-dependent fashion (Fig.
5B). Then, we used the Akt inhibitor (MK2206) to check the role
of Akt in the proliferative effect of CKIP-1. We found that MK2206
significantly reduced p-Akt and p-GSK-3β expression in Raji and
OCI-Ly8 cells (Fig. 5C). Notably,
promotion of cell proliferation mediated by CKIP-1 was reversed by
MK2206 in both Raji and OCI-Ly8 cells (Fig. 5D). Flow cytometric assays were used
to investigate the effect of CKIP-1 on apoptosis. This showed that
there were higher proportions of apoptotic cells in
CKIP-1-siRNA-transfected cells compared with control-siRNA
transfected ones. Moreover, CKIP-1-siRNA-mediated apoptosis was
increased in cells treated with MK2206. As expected, overexpression
of CKIP-1 reduced apoptosis; however, anti-apoptotic effects of
CKIP-1 were abrogated in MK2206-treated cells (Fig. 5E). All these data suggested that
knockdown of CKIP-1 promoted cell proliferation and inhibited
apoptosis by enhancing phospho-Akt expression.
CKIP-1 reverses CAM-DR via the Akt
pathway
Previous data suggested that CKIP-1 played an
important role in the CAM-DR phenotype. It has been reported that
the Akt signaling is the major pathway controlling cell growth and
survival, and elevated phosphorylated Akt levels could potentially
be causative for resistance to chemotherapy (17–19).
Thus, we verified the role of Akt in cell adhesion model. Western
blot analysis showed that the phosphorylation levels of Akt (Ser473
and Thr308) was more obvious in cells adhered to FN or HS-5 cells
than cells cultured in suspension, suggesting that Akt signaling
pathways promoted drug resistance (Fig.
6A). Thus, we suspected that CKIP-1 affected the CAM-DR by
regulating the activity of Akt signaling pathways. Then, to
determine whether CKIP-1 regulated CAM-DR phenotype via the Akt
pathway, Raji and OCI-Ly8 cells were treated with MK2206 or DMSO
(control). CCK-8 assays showed that knockdown of CKIP-1 in OCI-Ly8
and Daudi cell adhesion to FN or HS-5 cells significantly increased
cell viability, but the protective effect mediated by knockdown of
CKIP-1 was clearly reversed in MK2206-treated cells (Fig. 6B and C). All these data indicated
that CKIP reversed CAM-DR via Akt pathway.
Discussion
Non-Hodgkins lymphoma (NHL) comprises a
heterogeneous group of lymphoproliferative disorders containing
indolent as well as aggressive subtypes (1–3). The
incidence of NHL was increased in recent years and is still
associated with significant mortality. Despite intensive efforts in
developing new therapies, the emergence of clinical drug resistance
remains a barrier to successful treatment of lymphoma (6). Previous studies have shown that NHL
cells adhered to FN or stromal cells (HS-5 cells) confer a
multidrug-resistant phenotype and that disruption of cell
adhesion-mediated signaling may increase the efficacy of
chemotherapy drugs (20,21).
CKIP-1, also known as PLEKHO1, the PH
domain-containing casein kinase 2 interacting protein-1, has been
identified to play an important role in regulating many processes
such as involved in tumor cell proliferation, muscle cell
differentiation and cell apoptosis (8). In many cancers, the expression of
CKIP-1 is markedly declining, including human osteosarcoma, colon
(11) and breast cancer (15). Knockdown of CKIP-1 in macrophages
can shorten the G1 phase and accelerate cell proliferation
(13). In human colon cancer,
CKIP-1 functions as a potential tumor suppressor and scaffold
molecule that may modify and regulate the activities of several
signaling pathways, including PI3K/mTOR and TGF-β/BMP (11). Furthermore, CKIP-1 enhances the
sensitivity to chemotherapy drugs by targeting the Akt PH domain
and suppressing Akt kinase activity in human cancers (8). Although CKIP-1 has been reported to
reduce cell proliferation as a tumor suppressive gene in various
cancers (8,10,15,22),
the biological role of CKIP-1 in the formation and progression of
NHL remains unknown.
In the present study, we investigated the function
of CKIP-1 in NHL for the first time. Firstly, we demonstrated that
CKIP-1 was highly associated with NHL cell proliferation, and
knockdown of CKIP-1 could promote the proliferation of NHL cells
(Fig. 2C-F). Then, we determined
the mechanism underlying the effect of CKIP-1 on regulating cell
proliferation, and flow cytometric analysis revealed that knockdown
of CKIP-1 generate a decrease in the percentage of cells in the G1
phase, but more cells were found in the G1 phase in the presence of
myc-CKIP-1 (Fig. 3A and B). These
findings revealed that the arrest in G1 phase might be the major
reason for its inhibition of proliferation after CKIP-1 was
overexpressed in NHL cells. Moreover, we detected the expression
levels of mRNA transcripts encoding cell cycle-regulatory proteins
in NHL cell. OCI-Ly8 and Raji cells transfected with CKIP-1-siRNA
expressed more c-Myc, cyclin D1 mRNA and cyclin E mRNA compared
with control-siRNA transfected ones (Fig. 3C).
It has been widely demonstrated that CAM-DR through
direct cell contact and adhesion may be a major cause of tumor cell
drug resistance in hematologic malignancies (23–25).
The present study showed that adhesion to HS-5 cells or FN
decreased CKIP-1 expression, and the depressed levels of CKIP-1
significantly promote the proliferation of NHL cells (Fig. 2C-F). We found that knockdown of
CKIP-1 could promote CAM-DR phenotype in NHL (Fig. 4D and E). Therefore, the apparent
opposing roles of CKIP-1 suggest that, transient upregulation of
CKIP-1 prior to treatment with chemotherapy may have a therapeutic
benefit. However, how adhesion to FN or HS-5 cells influences
CKIP-1 expression, as well as the molecular mechanisms involved, is
still unclear and needs to be further elucidated.
For this reason, we elucidated the downstream target
of CKIP-1 in regulating NHL cell tumorigenesis. In this regard, a
previous study has identified that CKIP-1 interacts with Akt, and
suppresses Akt phosphorylation and decreases Akt kinase activity in
many human cancers (10). It has
been reported that the PI3K/Akt signaling is the major pathway
controlling cell growth and survival, and the elevated
phosphorylated Akt levels could potentially be causative for
resistance to chemotherapy (26,27).
Thus, we verified the role of PI3K/Akt in cell adhesion model.
Western blot analysis revealed that the phosphorylation levels of
Akt (Ser473 and Thr308) was more obvious in cells adhered to FN or
HS-5 cells than cells cultured in suspension, suggesting that
PI3K/Akt signaling pathways are involved in drug resistance
(Fig. 6A). Moreover, Raji and
OCI-Ly8 cells were transfected with myc-CKIP-1 in different doses.
We found that knockdown of CKIP-1 markedly upregulated the
phosphorylated Akt levels at Thr308 and Ser473 and the
phosphorylation of GSK-3β in a dose-dependent fashion (Fig. 5B). Consequently, we detected the
levels of apoptosis in CKIP-1 silencing cells and found that
knockdown of CKIP-1 decreased cell apoptosis in the cell adhesion
model, which can be reversed by Akt inhibitors (Fig. 6C and D). In summary, all data
suggested that knockdown of CKIP-1 could accelerate proliferation
and CAM-DR primarily through regulating the phosphorylation levels
of Akt in NHL.
In conclusion, the present study suggests that
CKIP-1 functions as a potential tumor suppressor and was highly
associated with cell proliferation and played an important role in
CAM-DR in NHL. Knockdown of CKIP-1 could accelerate the
proliferation of NHL cells and promote CAM-DR phenotype in NHL. In
the future, this may be an effective strategy for treatment of
NHL.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81372537,
81570187 and 81070400), the Nantong University Foundation of
Graduate Students of Science and Technology Innovation Project (no.
YKC15053), the Medical Innovation Team and the Leading Talent
Project of Jiangsu Province (LJ201136) and the Social Science and
Technology Innovation and Demonstration in Nantong-Clinical Medical
Science and Technology Project (HS2013067).
Glossary
Abbreviations
Abbreviations:
|
NHL
|
non-Hodgkins lymphoma
|
|
FBS
|
fetal bovine serum
|
|
CAM-DR
|
cell adhesion mediated drug
resistance
|
References
|
1
|
Harris NL, Jaffe ES, Stein H, Banks PM,
Chan JK, Cleary ML, Delsol G, De Wolf-Peeters C, Falini B, Gatter
KC, et al: A revised European-American classification of lymphoid
neoplasms: A proposal from the International Lymphoma Study Group.
Blood. 84:1361–1392. 1994.PubMed/NCBI
|
|
2
|
Maxwell SA and Mousavi-Fard S:
Non-Hodgkins B-cell lymphoma: Advances in molecular strategies
targeting drug resistance. Exp Biol Med (Maywood). 238:971–990.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shankland KR, Armitage JO and Hancock BW:
Non-Hodgkin lymphoma. Lancet. 380:848–857. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yagi K, Yamamoto K, Umeda S, Abe S, Suzuki
S, Onishi I, Kirimura S, Fukayama M, Arai A, Kitagawa M, et al:
Expression of multidrug resistance 1 gene in B-cell lymphomas:
Association with follicular dendritic cells. Histopathology.
62:414–420. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lwin T, Hazlehurst LA, Dessureault S, Lai
R, Bai W, Sotomayor E, Moscinski LC, Dalton WS and Tao J: Cell
adhesion induces p27Kip1-associated cell-cycle arrest
through down-regulation of the SCFSkp2 ubiquitin ligase
pathway in mantle-cell and other non-Hodgkin B-cell lymphomas.
Blood. 110:1631–1638. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lwin T, Zhao X, Cheng F, Zhang X, Huang A,
Shah B, Zhang Y, Moscinski LC, Choi YS, Kozikowski AP, et al: A
microenvironment-mediated c-Myc/miR-548m/HDAC6 amplification loop
in non-Hodgkin B cell lymphomas. J Clin Invest. 123:4612–4626.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mraz M, Zent CS, Church AK, Jelinek DF, Wu
X, Pospisilova S, Ansell SM, Novak AJ, Kay NE, Witzig TE, et al:
Bone marrow stromal cells protect lymphoma B-cells from
rituximab-induced apoptosis and targeting integrin α-4-β-1 (VLA-4)
with natalizumab can overcome this resistance. Br J Haematol.
155:53–64. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nie J, Liu L, He F, Fu X, Han W and Zhang
L: CKIP-1: A scaffold protein and potential therapeutic target
integrating multiple signaling pathways and physiological
functions. Ageing Res Rev. 12:276–281. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang L, Xing G, Tie Y, Tang Y, Tian C, Li
L, Sun L, Wei H, Zhu Y and He F: Role for the pleckstrin homology
domain-containing protein CKIP-1 in AP-1 regulation and apoptosis.
EMBO J. 24:766–778. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tokuda E, Fujita N, Oh-hara T, Sato S,
Kurata A, Katayama R, Itoh T, Takenawa T, Miyazono K and Tsuruo T:
Casein kinase 2-interacting protein-1, a novel Akt pleckstrin
homology domain-interacting protein, down-regulates PI3K/Akt
signaling and suppresses tumor growth in vivo. Cancer Res.
67:9666–9676. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nie J, Liu L, Xing G, Zhang M, Wei R, Guo
M, Li X, Xie P, Li L, He F, et al: CKIP-1 acts as a colonic tumor
suppressor by repressing oncogenic Smurf1 synthesis and promoting
Smurf1 autodegradation. Oncogene. 33:3677–3687. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vilk G, Saulnier RB, St Pierre R and
Litchfield DW: Inducible expression of protein kinase CK2 in
mammalian cells. Evidence for functional specialization of CK2
isoforms. J Biol Chem. 274:14406–14414. 1999.
|
|
13
|
Zhang L, Wang Y, Xiao F, Wang S, Xing G,
Li Y, Yin X, Lu K, Wei R, Fan J, et al: CKIP-1 regulates macrophage
proliferation by inhibiting TRAF6-mediated Akt activation. Cell
Res. 24:742–761. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Y, Liu F, Mao F, Hang Q, Huang X, He
S, Wang Y, Cheng C, Wang H, Xu G, et al: Interaction with cyclin
H/cyclin-dependent kinase 7 (CCNH/CDK7) stabilizes C-terminal
binding protein 2 (CtBP2) and promotes cancer cell migration. J
Biol Chem. 288:9028–9034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang L, Tie Y, Tian C, Xing G, Song Y,
Zhu Y, Sun Z and He F: CKIP-1 recruits nuclear ATM partially to the
plasma membrane through interaction with ATM. Cell Signal.
18:1386–1395. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Canton DA, Olsten ME, Niederstrasser H,
Cooper JA and Litchfield DW: The role of CKIP-1 in cell morphology
depends on its interaction with actin-capping protein. J Biol Chem.
281:36347–36359. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hennessy BT, Smith DL, Ram PT, Lu Y and
Mills GB: Exploiting the PI3K/AKT pathway for cancer drug
discovery. Nat Rev Drug Discov. 4:988–1004. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu T, Fang Y, Zhang H, Deng M, Gao B, Niu
N, Yu J, Lee S, Kim J, Qin B, et al: HEATR1 negatively regulates
Akt to help sensitize pancreatic cancer cells to chemotherapy.
Cancer Res. 76:572–581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Perna D, Karreth FA, Rust AG,
Perez-Mancera PA, Rashid M, Iorio F, Alifrangis C, Arends MJ,
Bosenberg MW, Bollag G, et al: BRAF inhibitor resistance mediated
by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc
Natl Acad Sci USA. 112:E536–E545. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hazlehurst LA, Damiano JS, Buyuksal I,
Pledger WJ and Dalton WS: Adhesion to fibronectin via β1 integrins
regulates p27kip1 levels and contributes to cell
adhesion mediated drug resistance (CAM-DR). Oncogene. 19:4319–4327.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lwin T, Hazlehurst LA, Li Z, Dessureault
S, Sotomayor E, Moscinski LC, Dalton WS and Tao J: Bone marrow
stromal cells prevent apoptosis of lymphoma cells by upregulation
of anti-apoptotic proteins associated with activation of NF-kappaB
(RelB/p52) in non-Hodgkins lymphoma cells. Leukemia. 21:1521–1531.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang L, Tang Y, Tie Y, Tian C, Wang J,
Dong Y, Sun Z and He F: The PH domain containing protein CKIP-1
binds to IFP35 and Nmi and is involved in cytokine signaling. Cell
Signal. 19:932–944. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li ZW and Dalton WS: Tumor
microenvironment and drug resistance in hematologic malignancies.
Blood Rev. 20:333–342. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang K, Jiang Y, Zheng W, Liu Z, Li H, Lou
J, Gu M and Wang X: Silencing of human
phosphatidylethanolamine-binding protein 4 enhances
rituximab-induced death and chemosensitization in B-cell lymphoma.
PLoS One. 8:e568292013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakagawa Y, Nakayama H, Nagata M, Yoshida
R, Kawahara K, Hirosue A, Tanaka T, Yuno A, Matsuoka Y, Kojima T,
et al: Overexpression of fibronectin confers cell adhesion-mediated
drug resistance (CAM-DR) against 5-FU in oral squamous cell
carcinoma cells. Int J Oncol. 44:1376–1384. 2014.PubMed/NCBI
|
|
26
|
Bai H, Li H, Li W, Gui T, Yang J, Cao D
and Shen K: The PI3K/AKT/mTOR pathway is a potential predictor of
distinct invasive and migratory capacities in human ovarian cancer
cell lines. Oncotarget. 6:25520–25532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu M, Qi Z, Liu B, Ren Y, Li H, Yang G
and Zhang Q: RY-2f, an isoflavone analog, overcomes cisplatin
resistance to inhibit ovarian tumorigenesis via targeting the
PI3K/AKT/mTOR signaling pathway. Oncotarget. 6:25281–25294. 2015.
View Article : Google Scholar : PubMed/NCBI
|