Introduction
Hepatocellular carcinoma (HCC) is the most common
primary liver tumor and the third most frequent cause of
cancer-related death worldwide (1).
Surgical resection and traditional chemotherapy are typical forms
of treatment for patients with HCC (2). Sorafenib (Nexavar) is the first and
only targeted therapy to clinically improve overall survival in
patients with advanced HCC, and has given hope to researchers for
effective agents to combat HCC (3),
but the clinical response is seriously limited by drug resistance.
To develop optimum strategy for overcoming sorafenib treatment
resistance is one of the major concerns in currently liver cancer
treatment. Long-lasting endeavors have been made in identifying
genes inducing chemoresistances in the past decades (4,5).
However, the link between specific gene and sorafenib treatment
resistance in liver cancer therapy remains unclear.
Non-coding RNAs (ncRNA) have recently been
implicated in hepatocarcinogenesis and tumor progression, represent
promising targets for cancer (6),
ncRNAs include short and long non-coding RNAs (7). Short ncRNAs have a length of under 200
nucleotides (nt) while long ncRNAs (lncRNAs) are more than 200 nt
in length, frequently ranging up to 100 kb (8). Recent studies estimated that the
number of lncRNAs in humans is approximately 15,000 and found that
most lncRNAs displayed tissue-specific expression patterns
(9). Increasing evidence identified
that lncRNAs epigenetically regulate the expression of genes
involved in fundamental cellular processes such as cell
proliferation, apoptosis, differentiation, and migration,
suggesting their potential oncogenic or tumor suppressing roles in
cancer development (10). The
TUC338 (transcribed ultra-conserved region 338) gene was first
identified as uc.338, along with 480 other ultra-conserved elements
in the human genome. Expression of this RNA gene has been found to
markedly increase in hepatocellular carcinoma (HCC) cells (11). Moreover, TUC338 expression
correlates with disease progression, and raises the possibility
that it may be involved in malignant transformation (12). It is also reported that lost
restoration of TUC338 in liver cancer cells inhibit their cell
proliferation and invasion (13),
indicating a functional role in liver cancer cell growth.
RAS proteins control many cellular processes,
including cell migration, proliferation, differentiation, and
survival. The RAS GTPase-activationg protein (RasGAP) gene RASAL1
has been demonstrated as a tumor suppressor gene which act as a
negative modulator of the RAS signaling pathway by catalyzing RAS
inactivation (14). RASAL1
expression is decreased in many tumors, including colorectal tumor
(15), thyroid cancer (16), gastric cancer (17), prostatic cancer and bladder cancer
(18). However, no data have been
reported on its association with chemoresistance. RASAL1 is
stimulated by increases in intracellular Ca2+ leading to
the attenuation of RAS activation and MAPK activity, contributing
to tumor progression through its weakened anti-RAS activity
(19). Activated RAS is involved
not only in tumor progression but also possibly in the development
of resistance of tumor cells to chemotherapy and to ionizing
radiation (20), RASAL1 might
inhibit tumor development and sensitivity to drug-therapy by
negatively regulating RAS activation.
Based on findings that the TUC338 and RASAL1
functionally regulate the tumor progression, we postulated that
TUC338 might play a role in the chemotherapy resistance of liver
cancer cells, which might be mediated by its regulation of the
expression of RASAL1. In the present study, we explored the
functional significance of TUC338/RASAL1 axis in the
sorafenib-resistance in liver cancer cells by in vitro and
in vivo studies.
Our results showed that elevated expression of
TUC338 was found in HCC tissues and cell lines, and knockdown
TUC338 in liver cancer cells inhibit the cell proliferation and
invasion ability accompanied with the overexpression of RASAL1.
Furthermore, knockdown TUC338 inhibited the RASAL1 pathway and
decreased the tumor growth genes, by increase the RASAL1
transcription. Silencing of TUC338 in HepG2/Sor cells could also
decrease the cell proliferation and eradicated its resistance to
sorafenib in vitro and in vivo. These findings
suggest that TUC338 might promote sorafenib-resistance in liver
cancer, which is mediated via its regulation of RASAL1 and this
might be a novel therapeutic target for sorafenib-resistant liver
cancer therapy.
Materials and methods
Samples
Twelve pairs of HCC samples and adjacent non-tumor
tissues were obtained from surgical specimens at department of
hepatobiliary surgery, the first affiliated hospital of Wenzhou
Medical University (Wenzhou, China) after informed consent. Clear
hepatocellular carcinoma was diagnosed histopathologically. All
these specimens were snap-frozen in liquid nitrogen after excision.
The study methodologies conformed to the standards set by the
Declaration of Helsinki. Informed consent was obtained from each
participant, collection and usage of all specimens were approved by
the local ethics committee.
Cell culture and reagents
The human hepatoma cell lines, HepG2, SMMC-7721,
BEK-7402, Hep3B, and Huh-7, and the liver cell line L02 were
purchased from the cell bank of Type Culture Collection at the
Chinese Academy of Sciences (Shanghai, China). The cells were
maintained in RPMI-1640 or DMEM supplemented with 10% fetal bovine
serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C
in 5% CO2 incubation. Sorafenib (BAY 43-9006) was
purchased from MedChem Express (Princeton, NJ, USA) and dissolved
in DMSO. The final DMSO concentration was <0.1%.
siRNA and transfection
For TUC338 knockdown experiments, siRNA was designed
and synthesized by GenePharma Co., Ltd (Shanghai, China), and the
sequences were as follows: si-TUC338: SenseSeq:
5′-CCACAGGACAGGUACAGCATT-3′, AntiSeq: 5′-UCAGUCUCCAGGGCUGUCATT-3′;
si-NC: SenseSeq: 5′-UUCUCCGAACGUGUACGUTT-3′, AntiSeq:
5′-ACGUGACACGUUCGGAGAATT-3′. HepG2, SMMC-7721 and Huh-7 cells were
plated at a density of 2×105 cells/well in 6-well
plates, overnight culture, cells were transfected with si-NC or
si-TUC338 by Lipo2000 (11668019, Life Technology) using 100 nM/well
siRNA in a 6-well plate following the manufacturer's instructions.
At 48 h after transfection, total protein and RNA samples were
prepared, proliferation assay, invasion assay, luciferase assay
were carried as described below.
Real-time PCR
Total RNA was extracted using TRIzol®
reagent (Invitrogen). DNaseI-treated RNA was used for first strand
cDNA synthesis using M-MLV reverse transcriptase (Promega) and
oligo (dT) 15 according to the manufacturer's protocols and 1 µl
cDNA samples were used for conventional PCR amplification.
Real-time quantitative PCR analysis was performed in a real-time
PCR system (StepOne, Applied Biosystems) and the expression levels
of TUC338 were normalized to 18s determined by a SYBR Green-based
comparative cycle threshold CT method. Real-time PCR primers were:
TUC338-F: 5′-GGTGAGAGGGGATGTTCAGT-3′, TUC338-R:
5′-TGGGTGAAATGAGGTTGGGG-3′; RASAL1-F:
5′-TGGATTTCTCTTCTTGCGATTCT-3′, RASAL1-R: 5′-TGTTGGTCCCGAAGGTCAA-3′;
VEGF-F: 5′-AGGGCAGAATCATCACGAAGT-3′, VEGF-R:
5′-AGGGTCTCGATTGGATGGCA-3′; GLUT-1-F: 5′-GGCCAAGAGTGTGCTAAAGAA-3′,
GLUT-1-R: 5′-ACAGCGTTGATGCCAGACAG-3′; MDR-1-F:
5′-TTGCTGCTTACATTCAGGTTTCA-3′, MDR-1-R:
5′-AGCCTATCTCCTGTCGCATTA-3′; 18s-F: 5′-CGCTTCCTTACCTGGTTGAT-3′,
18s-R: 5′-GAGCGACCAAAGGAACGATA-3′.
Western blotting
Total proteins were extracted with RIPA buffer
containing proteinase/phosphatase inhibitors (Thermo Fisher
Scientific, Cambridge, MA, USA). Thirty micrograms of cell lysate
and tumor tissue lysate were separated on 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and then
transferred to Immobilon-P transfer membrane (Millipore). Specific
anti-RASAL1 (ab170711, 1:1000 dilution), anti-VEGF (ab1316, 1:1000
dilution), anti-GLUT-1 (ab115730, 1:1000 dilution),
anti-P-glycoprotein (ab170904, 1:1000 dilution), anti-β-actin
(ab3280, 1:3000 dilution) primary antibodies (Abcam, USA) were
used, and HRP conjugated immunoglobulin was used as a secondary
antibody (Zen BioScience, Chengdu, China). Immunoblot analysis was
performed with the indicated antibodies and visualized with
Immobilon Western (Millipore), which were quantified using
densitometry image analysis software (Image Master VDS; Pharmacia
Biotech Inc.). Normalization was made against β-actin
expression.
In vitro cell growth assays
Cell counting
Viable cells were counted as previously described
(21). When the cells were cultured
to 70–80% confluence, sorafenib was added into the medium for
different concentrations at different time periods. All counts were
performed on triplicate wells and repeated in three independent
experiments and mean ± SEM of cell number was plotted against
culture duration of 8 days. For the sorafenib treatment assay, the
cell amount was confirmed to be comparable in the starting day for
each cell line by adjusting the cell amounts seeded according to
their doubling time.
MTT assay
Cells were seeded at 5×103 cells/well and
cultured under various concentration of sorafenib for different
time periods. Viable cells were determined by MTT assay as
previously described (22).
In vivo tumor study
Male nude mice (4–6 weeks, 18–20 g) were purchased
from the Model Animal Research Center of Nanjing University. The
animals were housed in a temperature- and humidity-controlled room
with a 12-h on-off light cycle and given free access to food and
water. To establish the exnograft mouse model, HepG2/Sor
sorafenib-resistant cancer cells were diluted with phosphate-buffer
saline (PBS) at a concentration of 2×107 cells/ml, and
200 µl HepG2/Sor cells were injected subcutaneously into the left
back limb of each mouse. Tumor volumes were determined every five
days after injection and calculated as previously described
(23). Mice with xenograft volume
>500 mm3 were treated with 4 mg/kg sorafenib (Sor)
intraperitoneally (i.p.) combined with intratumoral injection of
physiological saline twice a week (Sor+saline group), siNC
(Sor+siNC group) and siTUC338 (Sor+siTUC338 group) for four weeks.
Mice were sacrificed and tumors were dissected for real-time PCR
and western blot analysis. The animal study was carried out in
strict accordance with the recommendations in the guide for the
care and use of laboratory animals of the First Affiliated Hospital
of Wenzhou Medical University and Research Institute Animal Care
and Use Committee. All the protocols were approved by the First
Affiliated Hospital of Wenzhou Medical University and Research
Institute Animal Care and Use Committee (approval number 21040608).
Surgery was performed under sodium pentobarbital anesthesia, and
all efforts were made to minimize suffering.
Plasmid construction and luciferase assay
To determine whether the entire human RASAL1
3′-untranslated region (UTR) segment was amplified by PCR
homo-genomic DNA was used as a template. The PCR products were
inserted into the pGL3-Basic plasmid (Ambion). For luciferase
reporter assays, 1 µg of firefly luciferase reporter plasmid, 0.5
µg of β-galactosidase expression vector (Ambion), and equal amounts
(200 pmol) of siNC or siTUC338 were transfected into cells in
6-well plates. The β-galactosidase vector was used as a
transfection control. At 48 h after transfection, cells were
assayed using luciferase assay kits (Promega).
Statistical analysis
The results are expressed as mean ± SEM from at
least three independent experiments. Significance analysis of
normally distributed data were performed using two-tail Student's
t-test and P-values of <0.05 and 0.01 were considered
significant and highly significant, respectively.
Results
TUC338 expression is significantly
increased in HCC tissues and cell lines
To determine whether TUC338 was involved in
regulation of human HCC tumorigenesis, we first detected TUC338
levels in HCC tumor and adjacent non-tumor tissues, using real-time
PCR (n=12). As shown in Fig. 1A,
TUC338 expression was significantly upregulated in HCC samples
compared to normal adjacent liver tissue. We also analyzed TUC338
expression in liver cell line L02 and five human HCC cell lines
(HepG2, SMMC-7721, BEK-7402, Hep3B, and Huh-7). Consistently,
TUC338 was increased in all HCC cell lines examined compared to L02
cells (Fig. 1B). Taken together,
these findings suggested that TUC338 is upregulated in human
HCC.
Inhibition of TUC338 decreases cell
proliferation and invasion by activation of RASAL1
Functional role of TUC338 in liver cancer cells was
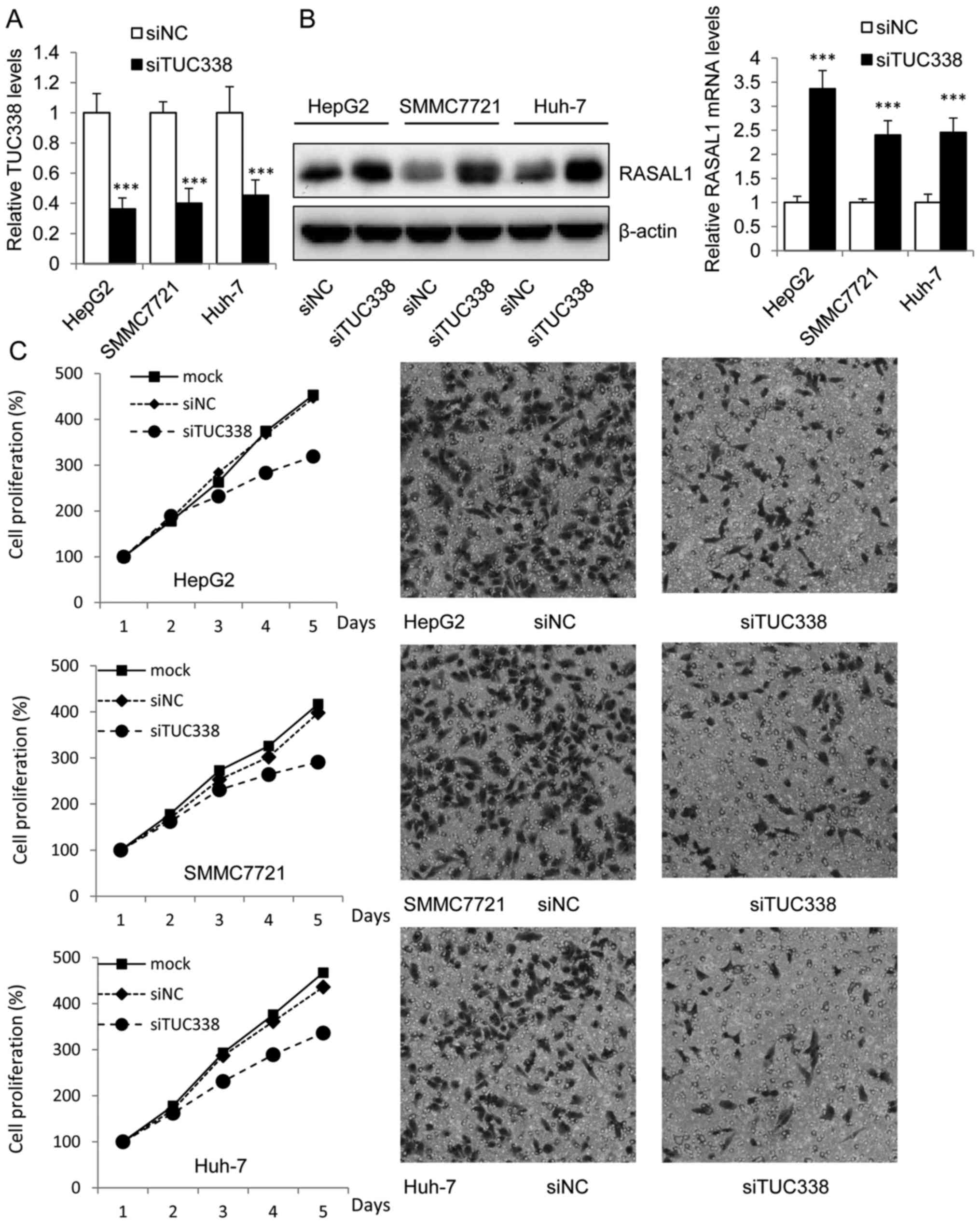
explored by siRNA knockdown TUC338 in HepG2, SMMC7721 and Huh-7
cells which showed higher basal expression levels of TUC338. It was
observed that the expression of TUC338 was significantly
downregulated 48 h after transfection with siTUC338 by Lipo2000 in
mRNA levels as compared with the siNC control (Fig. 2A). Moreover, the western blot assay
showed that transduction of siTUC338 in HCC cells could increase
the mRNA and protein levels of RASAL1 (Fig. 2B). These findings provide evidence
that TUC338 regulated the expression of RASAL1, regarded as tumor
suppressor in many tumors (14,24).
To establish whether siTUC338 plays a suppressing
role in HCC tumorigenesis, we analyzed cell viability using MTT
assay and invasion ability using Transwell assay in HCC cells
transfected with siTUC338. As shown in the left panel of Fig. 2C, knockdown of TUC338 markedly
reduced HCC cell viability in HepG2, SMMC7721 and Huh-7 cell lines.
Similarly, siTUC338 also significantly decreased incasion ability
of human HCC cells (Fig. 2C right
panel) in the three cell lines.
siTUC338 activates RASAL1 signaling
pathway and inhibits tumor growth genes by directly targeting
RASAL1
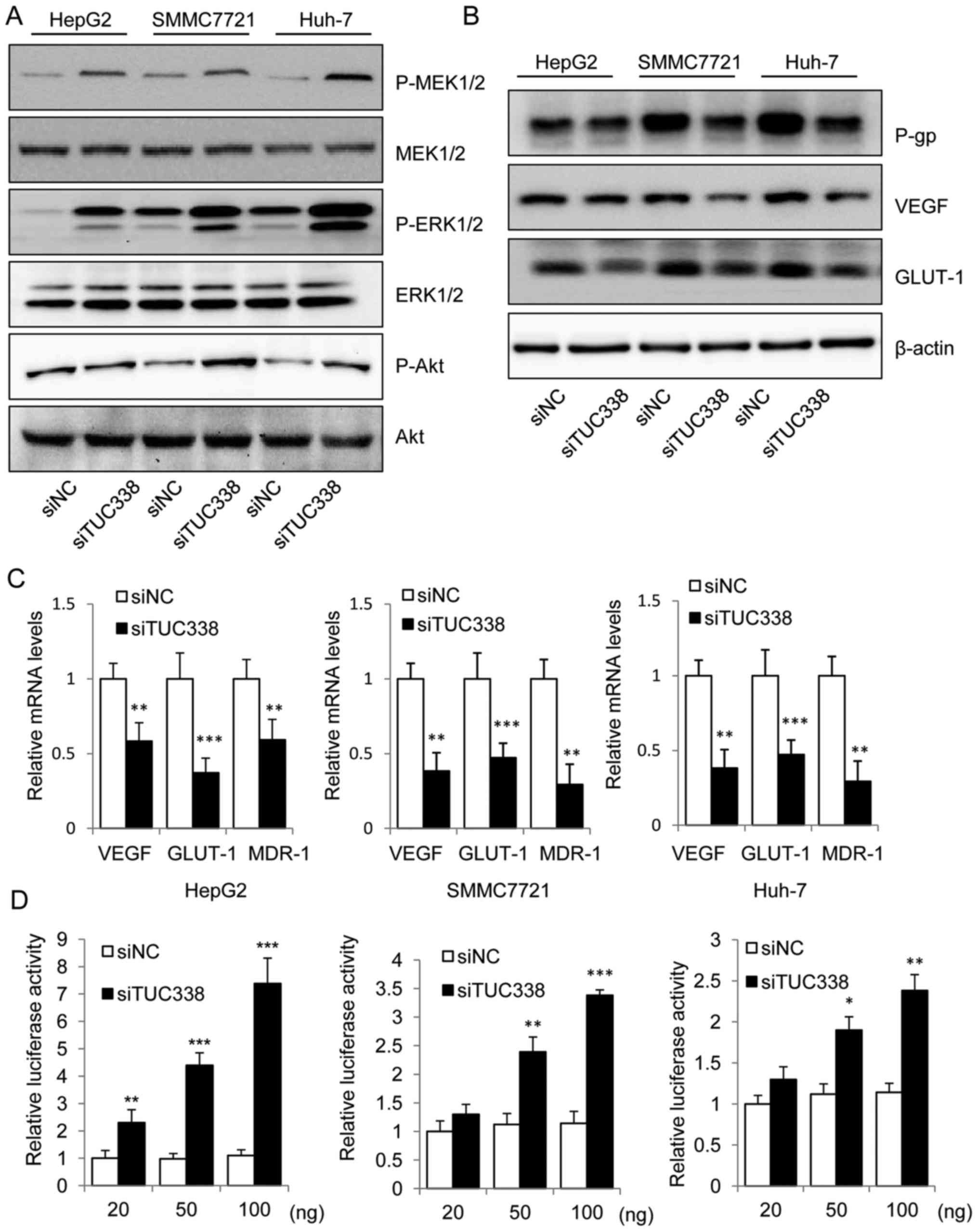
We next used real-time PCR and western blot to
examine whether TUC338 knockdown results in activation of RASAL1
signaling pathway. The immunoblot assay showed an increase in the
phospho-MEK1/2, phospho-ERK1/2 and phospho-Akt in TUC338 knockdown
HCC cells (Fig. 3A). Downregulated
expression of tumor growth genes vascular endothelial growth factor
(VEGF), glucose transporter 1 (GLUT-1), and multidrug resistance
gene (MDR1), produces P-glycoprotein; (P-gp) at the transcriptional
and the translational levels were also detected after TUC338
knockdown (Fig. 3B and C).
Moreover, to determine whether TUC338 could affect the
transcriptional activity of RASAL1, we co-transfected RASAL1
luciferase reporter plasmid with siNC or siTUC338 into HepG2,
SMMC7721 and Huh-7 cells. Of note, siTUC338 increased the relative
luciferase activity in a dose-dependent manner (Fig. 3D). Collectively, our data
demonstrate the functional link between TUC38 and the RASAL1
signaling pathway in human HCC cells.
Knockdown of TUC338 sensitizes HCC
cells to sorafenib
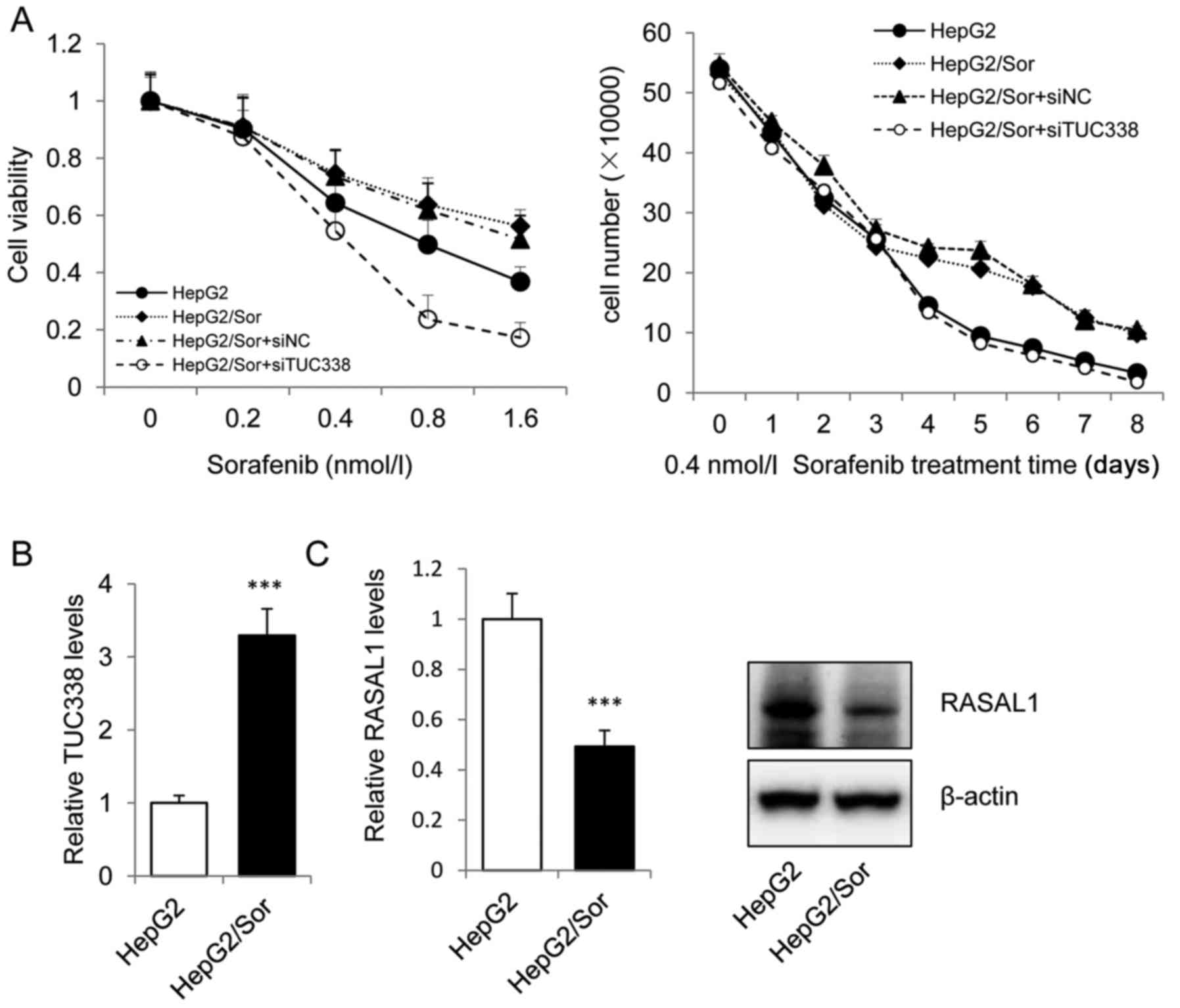
The sorafenib resistant subline HepG2/Sor was
gengrated by intermittent exposure of sensitive parental HepG2
cells to 30 ng/ml sorafenib in DMEM culture medium. We treated
HepG2, HepG2/Sor and siTUC338 or siNC transfected HepG2/Sor cells
with sorafenib and measured cell viability and counted the cell
number. Cell viability and cell number results showed that
transfected siNC HepG2/Sor cells are highly resistant to sorafenib
and that transfection with siTUC338 significantly reduced sorafenib
resistance (Fig. 4A). Next, we
investigated the mRNA and protein levels of TUC338 and RASAL1 in
HepG2/Sor cells using real-time PCR and western blot assay. As
shown in Fig. 4B and C, TUC338 was
overexpressed and RASAL1 was significantly downregulated both in
mRNA and protein levels in HepG2/Sor. Our data revealed that
knockdown of TUC338, an lncRNA which was elevated in HCC tissues
and cell lines, could sensitizes HCC cells to sorafenib by
upregulation of RASAL1, a major tumor suppressor.
Knockdown TUC338 helps to increase
sorafenib treatment response in sorafenib-resistant in vivo via
increasing the expression of RASAL1
Since lncRNA-TUC338 is capable of regulating RASAL1
expression in vitro, we next used the exnograft tumor nude
mouse model to investigate the effect of endogenous TUC338 on
RASAL1. After 4 weeks of treatment, the mean tumor volume in the
Sor+siTUC338 group was statistically lower compared to that in
Sor+siNC group and Sor group (Fig.
5A).
The TUC338 levels in tumor tissues were evaluated by
real-time PCR. After intratumoral injection with siTUC338, the
TUC338 was significantly decreased in the Sor+siTUC338 group
compare with the siNC and physiological saline control group
(Fig. 5B).
To understand the functions of TUC338 on the
sorafenib treatment by regulating the expression of RASAL1, RASAL1
mRNA and protein levels were valued. Real-time PCR and western
blots showed that the expression of RASAL1 both in mRNA and protein
levels were upregulated after intratumoral injection with siTUC338
(Fig. 5C).
Discussion
Resistance to the treatment of sorafenib is one of
the major obstacles in current metastatic liver cancer chemotherapy
(25,26). Increasing evidence indicates that
targeting lncRNA is a promising strategy to improve the efficacy of
treatment (27). Although great
endeavors have been made, more and more lncRNAs have been
discovered as effective chemotherapy inducers or inhibitor, which
may be attributed to the complexity of the development of
chemotherapy resistance.
To this end, we focused on TUC338, an lncRNA which
is overexpressed in liver cancer and may act as a tumor inducer, to
illustrate the function of lncRNA in the development process of
chemoresistance in liver cancer in vitro and in vivo.
Small interfering RNA (siRNA) was used to decrease the expression
of TUC338 in HCC cell lines, and its inhibition effect in the
proliferation and invasion ability was observed, consistent with
previous studies. Noteworthy, the downregulation of TUC338 was
accompanied with the upregulation of RASAL1, the activation of
RASAL1 pathway and the inhibition of tumor growth genes and
proteins. Luciferase assay showed that siTUC338 increased the
relative RASAL1 promoter luciferase activity in a dose-dependent
manner, and demonstrated the direct functional link between TUC38
and the RASAL1.
As a tumor suppressor, RASAL1 was found to inhibit
cancer progression and as a negative modulator of the RAS signaling
pathway by catalyzing RAS inactivation. It was evidenced that
enforced rexpression of RASAL1 in gastric cancer cells could
suppress cell proliferation and the transformation ability through
RAS/ERK signaling pathway (17,24),
while activated RAS is involved not only in tumor progression but
also possibly in the development of resistance of tumor cells to
chemotherapy and ionizing radiation, suggesting targeting this
oncogenic protein may be a potential strategy for the treatment of
chemoresistance. To this end, a sorafenib-resistant liver cancer
cell line, HepG2/Sor, was established, which could simplify and
better mimic the development process of chemoresistance in liver
cancer in vitro. We observed decreased expression of RASAL1
in HepG2/Sor at both mRNA and protein level as compared to that in
its parental cell line. More interestingly, this downregulation is
concomitant with increased expression of TUC338, which is evidenced
as a direct repressor of RASAL1.
Based on in vitro evidence that TUC338 and
RASAL1 might be involved in sorafenib-resistance in liver cancer
cells, the in vivo efficacy of inhibition of TUC338 in
sorafenib treatment were further explored. Our findings that
enhanced tumor growth inhibition efficacy induced by sorafenib
combined with intratumoral delivering siTUC338 accompanied with
overexpression of RASAL1 in tumor xenografts indicates that the
TUC338/RASAL1 axis could be a potential therapeutic target for
current liver cancer therapy.
The data obtained in the present study suggest that
the TUC338/RASAL1 axis might play an essential role in
sorafenib-resistance of liver cancer cells, siTUC338 was able to
inhibit the RASAL1 pathway and tumor growth genes by directly
targeting RASAL1 3′-UTR. Knockdown of TUC338 in HepG2/Sor could
sensitize its reaction to the treatment of sorafenib, which was
accompanied by the increased expression of RASAL1 both in
vivo and in vivo. Suggesting the signaling cohort could
serve as a novel therapeutic target for the treatment of
chemotherapy resistant liver cancer.
Acknowledgements
This work was supported by the Natural Science
Foundation of China (81201953) and the Natural Science Foundation
of Zhejiang Province (Y2090538).
References
|
1
|
Hernandez-Gea V, Toffanin S, Friedman SL
and Llovet JM: Role of the microenvironment in the pathogenesis and
treatment of hepatocellular carcinoma. Gastroenterology.
144:512–527. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
de Lope CR, Tremosini S, Forner A, Reig M
and Bruix J: Management of HCC. J Hepatol. 56:(Suppl 1). S75–S87.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Spinzi G and Paggi S: Sorafenib in
advanced hepatocellular carcinoma. N Engl J Med. 359:2497–2498.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Glackin CA: Targeting the Twist and Wnt
signaling pathways in metastatic breast cancer. Maturitas.
79:48–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Noguchi K, Katayama K and Sugimoto Y:
Human ABC transporter ABCG2/BCRP expression in chemoresistance:
Basic and clinical perspectives for molecular cancer therapeutics.
Pharm Genomics Pers Med. 7:53–64. 2014.
|
|
6
|
Parasramka MA, Maji S, Matsuda A, Yan IK
and Patel T: Long non-coding RNAs as novel targets for therapy in
hepatocellular carcinoma. Pharmacol Ther. 161:67–78. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Birney E, Stamatoyannopoulos JA, Dutta A,
Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis
ET, Thurman RE, et al: Children's Hospital Oakland Research
Institute: Identification and analysis of functional elements in 1%
of the human genome by the ENCODE pilot project. Nature.
447:799–816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Derrien T, Johnson R, Bussotti G, Tanzer
A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG,
et al: The GENCODE v7 catalog of human long noncoding RNAs:
Analysis of their gene structure, evolution, and expression. Genome
Res. 22:1775–1789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Prensner JR and Chinnaiyan AM: The
emergence of lncRNAs in cancer biology. Cancer Discov. 1:391–407.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Braconi C, Valeri N, Kogure T, Gasparini
P, Huang N, Nuovo GJ, Terracciano L, Croce CM and Patel T:
Expression and functional role of a transcribed noncoding RNA with
an ultraconserved element in hepatocellular carcinoma. Proc Natl
Acad Sci USA. 108:786–791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Braconi C and Patel T: Non-coding RNAs as
therapeutic targets in hepatocellular cancer. Curr Cancer Drug
Targets. 12:1073–1080. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
George J and Patel T: Noncoding RNA as
therapeutic targets for hepatocellular carcinoma. Semin Liver Dis.
35:63–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu D, Yang C, Bojdani E, Murugan AK and
Xing M: Identification of RASAL1 as a major tumor suppressor gene
in thyroid cancer. J Natl Cancer Inst. 105:1617–1627. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ohta M, Seto M, Ijichi H, Miyabayashi K,
Kudo Y, Mohri D, Asaoka Y, Tada M, Tanaka Y, Ikenoue T, et al:
Decreased expression of the RAS-GTPase activating protein RASAL1 is
associated with colorectal tumor progression. Gastroenterology.
136:206–216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ngeow J and Eng C: RASAL1 in thyroid
cancer: Wisdom from an old foe. J Natl Cancer Inst. 105:1597–1599.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen H, Cheng Z-Y, Pan Y, Wang Z, Liu Y
and Zhang J-Q: RASAL1 influences the proliferation and invasion of
gastric cancer cells by regulating the RAS/ERK signaling pathway.
Hum Cell. 27:103–110. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kolfschoten IG, van Leeuwen B, Berns K,
Mullenders J, Beijersbergen RL, Bernards R, Voorhoeve PM and Agami
R: A genetic screen identifies PITX1 as a suppressor of RAS
activity and tumorigenicity. Cell. 121:849–858. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Walker SA, Kupzig S, Bouyoucef D, Davies
LC, Tsuboi T, Bivona TG, Cozier GE, Lockyer PJ, Buckler A, Rutter
GA, et al: Identification of a Ras GTPase-activating protein
regulated by receptor-mediated Ca2+ oscillations. EMBO
J. 23:1749–1760. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Loriot Y, Mordant P, Deutsch E, Olaussen
KA and Soria J-C: Are RAS mutations predictive markers of
resistance to standard chemotherapy? Nat Rev Clin Oncol. 6:528–534.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li W, Zhai B, Zhi H, Li Y, Jia L, Ding C,
Zhang B and You W: Association of ABCB1, β tubulin I, and III with
multidrug resistance of MCF7/DOC subline from breast cancer cell
line MCF7. Tumour Biol. 35:8883–8891. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Febriansah R, Putri DD, Sarmoko Nurulita
NA, Meiyanto E and Nugroho AE: Hesperidin as a preventive
resistance agent in MCF-7 breast cancer cells line resistance to
doxorubicin. Asian Pac J Trop Biomed. 4:228–233. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tomayko MM and Reynolds CP: Determination
of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother
Pharmacol. 24:148–154. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qiao F, Su X, Qiu X, Qian D, Peng X, Chen
H, Zhao Z and Fan H: Enforced expression of RASAL1 suppresses cell
proliferation and the transformation ability of gastric cancer
cells. Oncol Rep. 28:1475–1481. 2012.PubMed/NCBI
|
|
25
|
Villanueva A and Llovet JM: Second-line
therapies in hepatocellular carcinoma: Emergence of resistance to
sorafenib. Clin Cancer Res. 18:1824–1826. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhai B and Sun X-Y: Mechanisms of
resistance to sorafenib and the corresponding strategies in
hepatocellular carcinoma. World J Hepatol. 5:345–352. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu W, Qiao Y, Tang X, Ma L, Wang Y, Zhang
X, Weng W, Pan Q, Yu Y, Sun F, et al: Tumor suppressor long
non-coding RNA, MT1DP is negatively regulated by YAP and Runx2 to
inhibit FoxA1 in liver cancer cells. Cell Signal. 26:2961–2968.
2014. View Article : Google Scholar : PubMed/NCBI
|