Introduction
Ovarian cancer is the most severe gynecologic
malignancy, causing 114,000 deaths a year globally. In the USA
alone, an estimated 23,000 women are diagnosed with ovarian cancer
each year and the 5-year survival rate is merely 30% (1). In the United States, ovarian cancer
represents 3% of all the new cancer cases in women, and accounts
for 5% of all the cancer deaths (2). The high mortality is partly due to the
frequent resistance of ovarian cancer to chemotherapy regimens.
Paclitaxel combined with platinum remains to be the first line
chemotherapy for ovarian cancer. Paclitaxel is a small molecule
cytotoxin targeting tubulin and has strong cytostatic and apoptotic
effects. Unfortunately, while most patients initially respond to
this combined chemotherapy, the majority of these (up to 75%) will
eventually relapse within 18 months with many having drug resistant
disease (3). Ovarian cancer cells
develop drug resistance through different pathways depending on the
drug used (4). Multiple mechanisms
can mediate the development of paclitaxel resistance, including
changes in: i) the regulation or repair of the primary target of
the drug (DNA, microtubule); ii) drug retention (increased efflux
or decreased uptake); iii) drug inactivation or sequestration; and
iv) signaling pathways that affect cell cycle/apoptosis. Paclitaxel
is known to be transported by the ATP-dependent efflux pump
P-glycoprotein (multidrug resistance, MDR) and upregulation of MDR
has been associated with clinical drug resistance to various agents
(5,6).
There is an imperative need for the development of
new treatment modalities to improve the management of ovarian
cancer patients. Switch to alternative drugs with different
therapeutic mechanisms is one strategy to overcome the resistance
against the presently used drugs. However, limited success has been
achieved with the use of second line chemotherapy following the
recurrence of ovarian cancer or the resistance to the first line
drugs (7). This failure is often
caused by the activation of ‘generic’ resistance mechanism against
multiple drugs sharing a specific feature. Rationalized design and
targeted chemotherapy using modified drugs equipped with new
features to avoid the resistance of cancer cells may potentially
enhance the drug efficacy and reduce the toxicity of cancer
therapies.
SST is a cyclic polypeptide hormone that is found in
most human organs and tissues. SST has a broad range of cellular
functions such as inhibition of secretion and blocking of cell
proliferation and cell survival (8,9).
Natural somatostatin (SST) has limited clinical applications
because of its low selectivity and short half-life. However,
somatostatin analogue (SSTA) is widely applied and has been shown
to have more powerful effects and a longer half-life. It has been
shown that SSTA is able to inhibit the proliferation of
neuroendocrine tumors in vitro as well as tumor growth in
vivo (10–14). The specific somatostatin receptor
(SSTR), with five subtypes, mediates the functions of SSTA. Two or
more receptor subtypes, particularly SSTR2 are often detected in
ovarian cancers (15,16) and most types of other tumors
(17,18). It is known that SSTR2 mediates the
inhibition of cell proliferation via the activation of Ras-, Rapl-
and B-Raf-dependent extracellular signal-regulated kinase 2 (Erk2)
(11,12,19).
Octreotide (OCT) is the most widely used SSTA in clinical
applications. OCT was found to exhibit the highest binding affinity
to SSTR2 and subsequently inhibit the activity of tyrosine
phosphatase and the proliferation of SSTR2-expressing cells
(20,21). In previous studies, we detected the
expression of SSTR2 in SKOV3/DDP by quantitative PCR and showed
that OCT could inhibit ovarian cancer proliferation and promote
apoptosis via the cell surface SSTR2. Furthermore, OCT could
reverse cisplatin resistance through inhibition of MRP2, EGFR and
GST-π expressions (22,23).
The mechanisms by which SST and SSTA enhance the
paclitaxel sensitivity of resistant ovarian cancer cells remain
unclear. OCT might be of practical value in developing tumor
tracers and in serving as a carrier of cytotoxic antitumor drugs.
The antitumor activity of paclitaxel relies on its capability of
promoting tubulin assembly into microtubules and the resultant
interference with the G2-M transition of cell cycle (24,25).
Huang et al (26) coupled
paclitaxel to SSTA and showed that the conjugate displayed an
increased cytotoxicity in vitro. Sun et al (27) and Shen et al (28) reported that paclitaxel-octreotide
conjugate (POC) could enhance tumor growth inhibition with reduced
toxicity in non-small cell lung cancer patients in comparison to
unconjugated paclitaxel. The above-mentioned evidence suggests the
conjugate triggers tumor cell apoptosis mediated by SSTRs and is
exclusively toxic to SSTR-expressing cells. Thus, the conjugate
could be less toxic to low-SSTR-expressing cells compared with free
paclitaxel.
In the present study, we prepared the POC and
investigated its function and mechanism for the reversal of drug
resistance in a paclitaxel-resistant ovarian cancer cell line. The
findings shed new light on the mechanisms of drug resistance and
may provide useful information for the development of better
treatment approach for ovarian cancer patients.
Materials and methods
The synthesis of POC
Direct synthesis of OCT acetic acid and paclitaxel
succinic acid derivatives was prepared for target products.
Paclitaxel of 200 mg and succinic anhydride of 300 mg were dried in
vacuum for 5 h, dissolved in 5 ml dry pyridine, and mixed for
reaction at 30°C for 24 h. The reaction products were re-dissolved
in 10 ml of acetone and the paclitaxel succinyl anhydride was
extracted from solid precipitation in conditions of drying and
reduced pressure, followed by adding and stirring with 10 ml of
water dropwise. Paclitaxel succinyl anhydride of 25 mg, SDPP
(N-hydroxysuccinimido diphenyl phosphate) of 30 mg and
triethylamine of 30 mg were dissolved in 0.5 ml anhydrous
acetonitrile with stirring overnight at room temperature. The
preliminary product mixture was followed by vacuum concentration
process and then re-dissolved into ethyl acetate. Finally, the
target product was successfully recovered by washing and drying
process.
Cell culture
Human ovarian cancer cell line A2780 (Institute of
Biochemistry and Cell Biology, Chinese Academy of Sciences,
Shanghai, China) and A2780/Taxol (Bogoo Biotechnology, Co., Ltd.,
Shanghai, China) were cultured at 37°C, 5% CO2
atmosphere and 90% humidity, in RPMI-1640 medium (Gibco, Carlsbad,
CA, USA) with 10% fetal bovine serum (FBS; Invitrogen, Waltham, MA,
USA). The cells were passaged every 2–3 days using 0.25% trypsin
(Sigma-Aldrich, Schnelldorf, Germany). The log-phase cells were
collected for further experiment.
Confocal microscopy
To evaluate the targeted binding of POC to SSTR2
positive cells, we observed the internalization of
fluorescein-labeled POC into A2780/Taxol cells at different times.
A2780/Taxol cells cultured with fluorescein-labeled POC (10
nmol/ml) were detected by confocal microscope (Olympus FluoView™
FV1000; Olympus, Tokyo, Japan) at 30 min, 2 h and 8 h when after
preparation of phosphate-buffered saline (PBS) buffer washing 3
times.
Cell proliferation assay
A2780/Taxol cells (Bogoo Biotechnology) in the
log-phase were seeded in each well of the 96-well culture plates
and cultured at 37°C under a 5% CO2 atmosphere for 24 h.
The cells were incubated in 100 µl of medium with paclitaxel (0,
1.25, 2.5, 5, 10 and 20 nmol/ml), OCT (0, 1.25, 2.5, 5, 10 and 20
nmol/ml), or POC (0, 1.25, 2.5, 5, 10 and 20 nmol/ml). At different
time-points, cells were treated with 10 µl of the Cell Counting
kit-8 (CCK-8; Dojindo Laboratories, Kumamoto Japan) for 3 h.
Absorbance (A) was measured on an enzyme-linked immunosorbent assay
plate reader. The inhibition rate was calculated using the
following formula: Cell proliferation inhibition rate = (average of
value A from the control group - the average of value A from the
experimental group)/(average of value A from the control group -
average of value A from blank controller) × 100%. Resistance index
was calculated with the following formula: IC50 of
resistant A2780 cells/IC50 of parental A2780 cells. All
experiments were repeated in triplicate and more than three wells
were used for each treatment.
Detection of cell apoptosis
The experiment contained four groups comprising the
control, paclitaxel (10 nmol/ml), OCT (10 nmol/ml) and POC (10
nmol/ml). Following treatment for 36 h, cell apoptosis was examined
using the Annexin V-FITC/PI staining kit (Nanjing KeyGen Biotech,
Co., Ltd., Nanjing, China) according to the instructions provided
by the manufacturer. Flow cytometry (FACSVantage SE; BD
Biosciences, San Jose, CA, USA) was performed and apoptotic cells
were counted for each group of treatment.
Immunocytochemistry
The cells cultured on coverslips in 6-well plates
were fixed in 4% paraformaldehyde for 30 min, washed with PBS for 5
min and permeabilized with Triton X-100 (Sigma-Aldrich). The cells
were incubated with 10% goat serum for 20 min for blocking. Primary
rabbit monoclonal antibody (anti-SSTR2, 1:100; Abcam) was added and
the incubation continued overnight at 4°C in a humidified chamber.
After washing with PBS, HRP-labeled secondary antibody was applied
for 30-min incubation. Coverslips were immersed in freshly prepared
DAB solution (Dako Denmark A/S, Glostrup, Denmark) for color
development. Cells were counterstained with hematoxylin for 10 min
and microscopic observation was performed for the detection of
SSTR2 expression.
Real-time PCR
Total RNA was extracted from A2780/Taxol cells
treated by conjugate for 48 h with the use of TRIzol reagents. RNA
concentration was measured on a UV spectrophotometer based on the
absorbance values at 260 and 280 nm. cDNA was synthesized using 1
µg of total RNA according to the instructions provided in the
RT-PCR kit (Takara). Designation and sequences of PCR primers
(Houzai Co., Tokyo, China) are provided in Table I. Real-time PCR was performed in a
LightCycler (Roche Applied Science) under the following conditions:
pre-80 denaturation at 94°C for 2 min, then denaturation at 94°C
for 45 sec, annealing at 56°C for 45 sec, extension at 72°C for 45
sec. Fold of difference relative to the reference gene (β-actin)
was determined by conversion of 2−∆∆CT. ∆∆CT =
(CTobjective gene - CTreference gene) of
experimental group - (CTobjective gene -
CTreference gene) of control group.
 | Table I.Primer sequences for the real-time
PCR reaction. |
Table I.
Primer sequences for the real-time
PCR reaction.
| Objective gene | Primer
sequence |
|---|
|
β-actin-Forward |
GATGACCCAGATCATGTTTGAG |
|
β-actin-Reverse |
AGGGCATACCCCTCGTAGAT |
| SSTR2-Forward |
CATTTATGTCATCCTCCGCTAT |
| SSTR2-Reverse |
TGATTGATGCCATCCACAGT |
| VEGF-Forward |
CAGAAGGAGGAGGGCAGAAT |
| VEGF-Reverse |
CACAGGATGGCTTGAAGATG |
| MDR1-Forward |
GCTGTCAGGTGCCATCAAT |
| MDR1-Reverse |
TGGAAGGGAGCGGTGTAA |
Western blot analysis
Following our previously established method
(29), A2780/Taxol cells treated by
conjugate for 48 h were lysed using modified RIPA lysis buffer (1%
NP-40, 0.25% deoxycholic acid, 50 mM Tris-HCl pH 7.4, 1 mM EDTA,
150 mM NaCl, 1 mM NaF, 1 g/ml leupeptin, 1 mM PMSF, 1 mM sodium
orthovanadate, 2 g/ml pepstatin and 1 g/ml aprotinin). Cell lysates
were boiled in the loading buffer (3.3% glycerin, 1% SDS, 20 mM
TRIS, pH 6.8, 23 mM β-mercaptoethanol freshly added and 0.4 mg/ml
bromophenol blue). Proteins were separated in precast gradient
SDS-PAGE (4–20%) and transferred to polyvinylidenfluorid (PDVF)
membranes (Bio-Rad Laboratories, Hercules, CA, USA), and then
incubated with specific primary antibodies for 2 h at room
temperature, followed by 1 h of incubation with appropriate
HRP-conjugated secondary antibodies. Western blot analyses were
performed by using primary antibodies against SSTR2 (1:1,000),
IGF-1 (1:600), VEGF (1:1,000). The antigen-antibody complexes in
western blot analysis were detected with an enhanced
chemiluminescence detection system (Amersham Biosciences,
Pittsburgh, PA, USA). Specific protein bands were visualized after
autoradiography. The intensity of each protein band was quantified
by using image analysis software and normalized against
corresponding β-actin that was detected by anti-β-actin antibody
(1:2,000).
Statistical analysis
Statistical analysis was performed using the SPSS
16.0 software. Data are expressed as the means ± SD. The ANOVA and
Student's t-test was used for comparison of the drug-treated and
drug-untreated controls. A P<0.05 was considered to indicate a
statistically significant result.
Results
Synthesis of POC
Direct synthesis of OCT acetic acid and paclitaxel
succinic acid derivatives was performed as described in Materials
and methods. As shown in Fig. 1,
the process consisted of sequential synthesis of the following four
chemicals: i) N-hydroxysuccinimido diphenyl phosphate; ii)
paclitaxel succinyl anhydride; iii) N-hydroxy paclitaxel succinyl
anhydride; and iv) POC.
SSTR2 expression in A2780/Taxol
cells
In order to confirm the SSTR2 expression in the cell
model, we performed immunocytochemistry using the specific antibody
against SSTR2. As shown in Fig. 2,
A2780/Taxol cell membranes display strong positive staining.
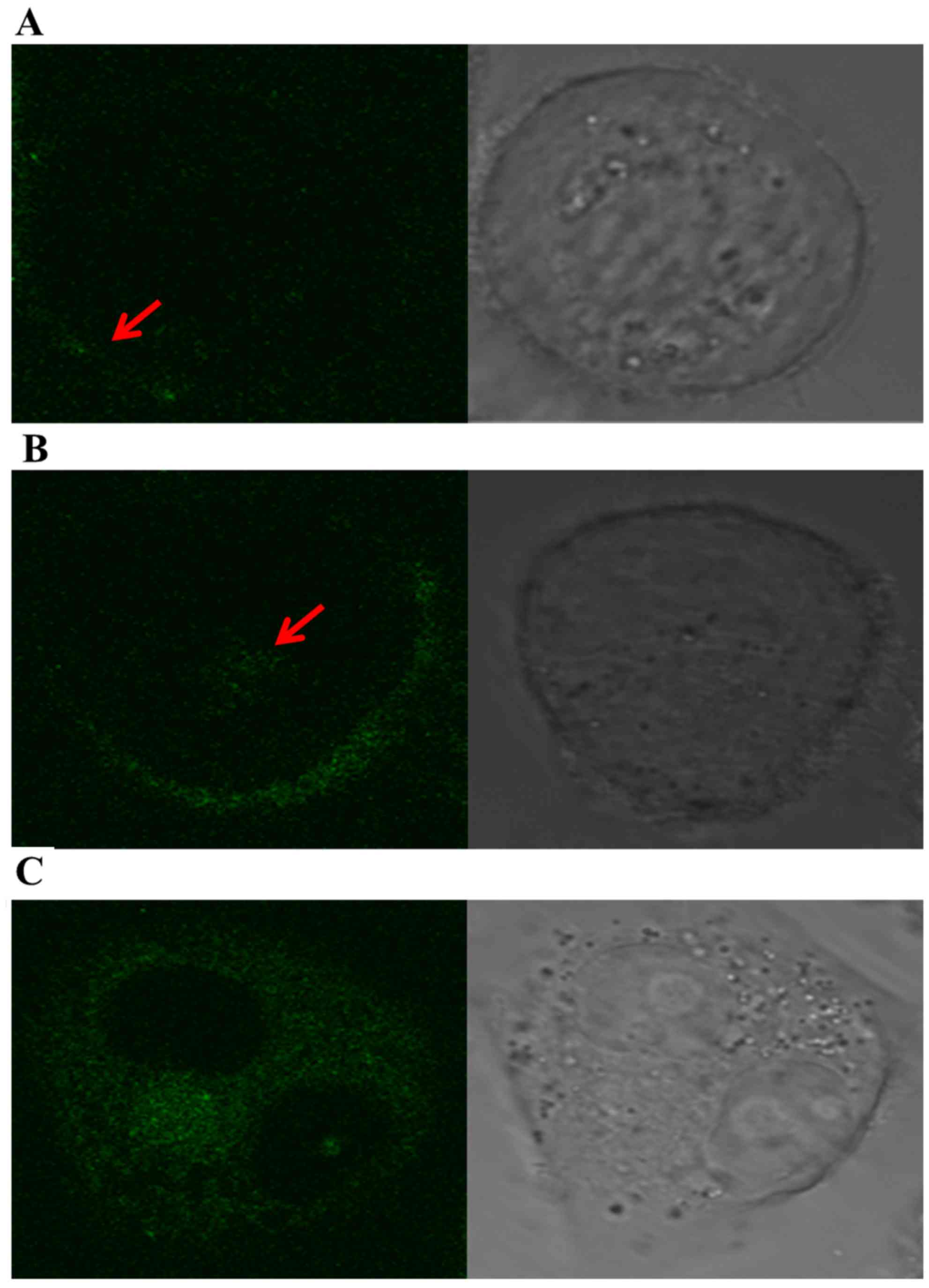
Internalization of fluorescein-labeled
POC in A2780/Taxol cells
The specific fluorescein were visible mainly along
the cell surface of the A2780/Taxol cells at 30 min of culture,
whereas part of the denser fluorescent grains appeared into the
cytoplasm at 2 h. Moreover, fluorescent grains were visibly
distributed through the cytoplasm, and many grains were
concentrated around cell nucleus at 8 h (Fig. 3). These results strongly suggested
that POC has a favorable and specific binding targeted to
SSTR2-positive cells, which resulted from the SSTR2-mediated
internalization in A2780/Taxol cells.
Effect of POC on A2780/Taxol cell
proliferation
The A2780/Taxol cells were round, cytoplasm-rich and
grew vigorously under normal conditions. Without treatment, cells
were transparent, spread evenly, and with smooth and complete edges
and had similar sizes and shapes. Following POC treatment for 24 h,
the number of adherent normal cells decreased, and the cell-cell
space became larger. Some cells appear to be condensed, darker and
displayed morphological features of apoptosis including shrinkage,
foaming and formation of apoptotic bodies.
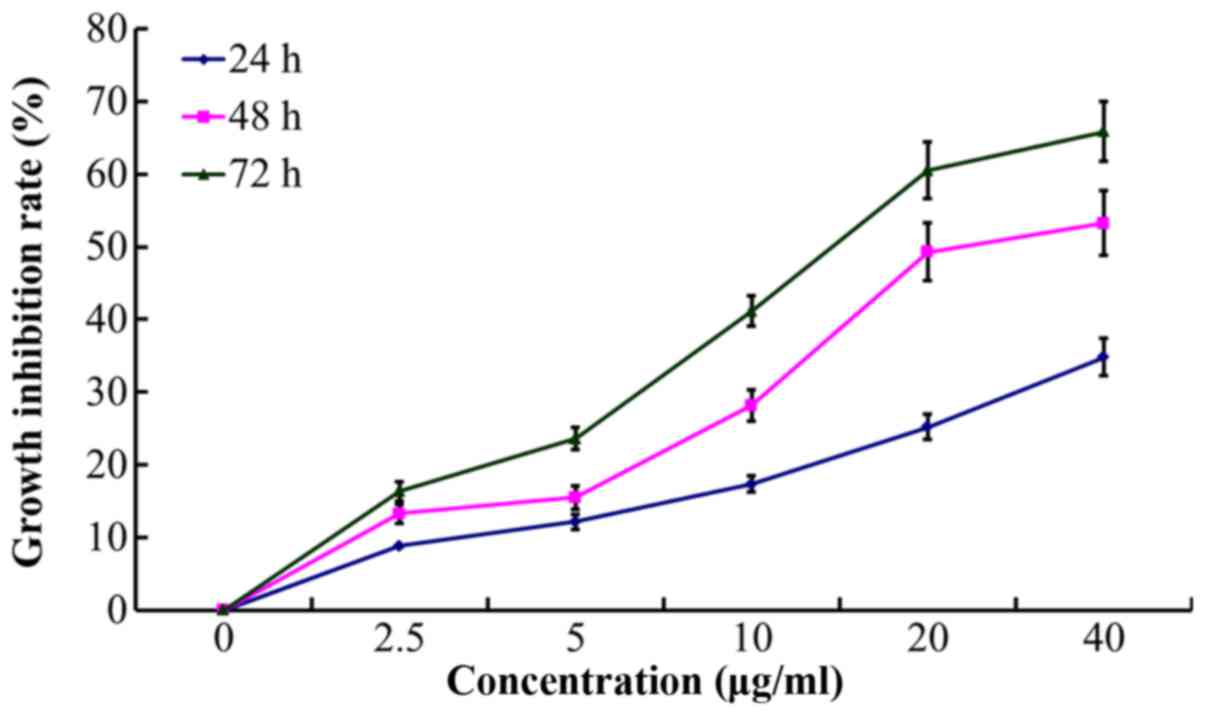
As the treatment time prolonged, cell growth became
slow and there were increased numbers of floating cells and cell
debris. According to the dose-effect curves of paclitaxel on
A2780/Taxol and A2780 cells, the calculated resistance index (RI)
of A2780/Taxol cell to paclitaxel was 28.33. POC exhibited an
enhanced inhibitory effect on A2780/Taxol cell proliferation, with
the calculated resistance index (RI) reaching 4.2 (Fig. 4). Notably, the POC inhibited
proliferation at the indicated time (24, 48 and 72 h) in a
concentration-dependent manner (P<0.05; Table II and Fig. 5).
| Table II.Inhibition rate of POC on A2780/Taxol
cell proliferation at the indicated concentration and time. |
Table II.
Inhibition rate of POC on A2780/Taxol
cell proliferation at the indicated concentration and time.
| Groups | POC (nmol/ml) | Time I (24 h) | Time II (48 h) | Time III (72
h) |
|---|
| 1 | 0 | 0 | 0 | 0 |
| 2 | 1.25a | 8.83±0.32 | 13.28±0.20 |
16.35±0.72f |
| 3 | 2.5a,b | 12.17±0.42 |
15.51±0.93f |
23.61±1.21f,g |
| 4 | 5.0a–c | 17.34±0.27 |
28.18±0.15f |
41.18±1.00f,g |
| 5 | 10.0a–d | 25.24±0.87 |
49.31±1.49f |
60.50±1.99f,g |
| 6 | 20.0a–e | 34.80±0.62 |
53.27±2.08f |
65.48±2.17f,g |
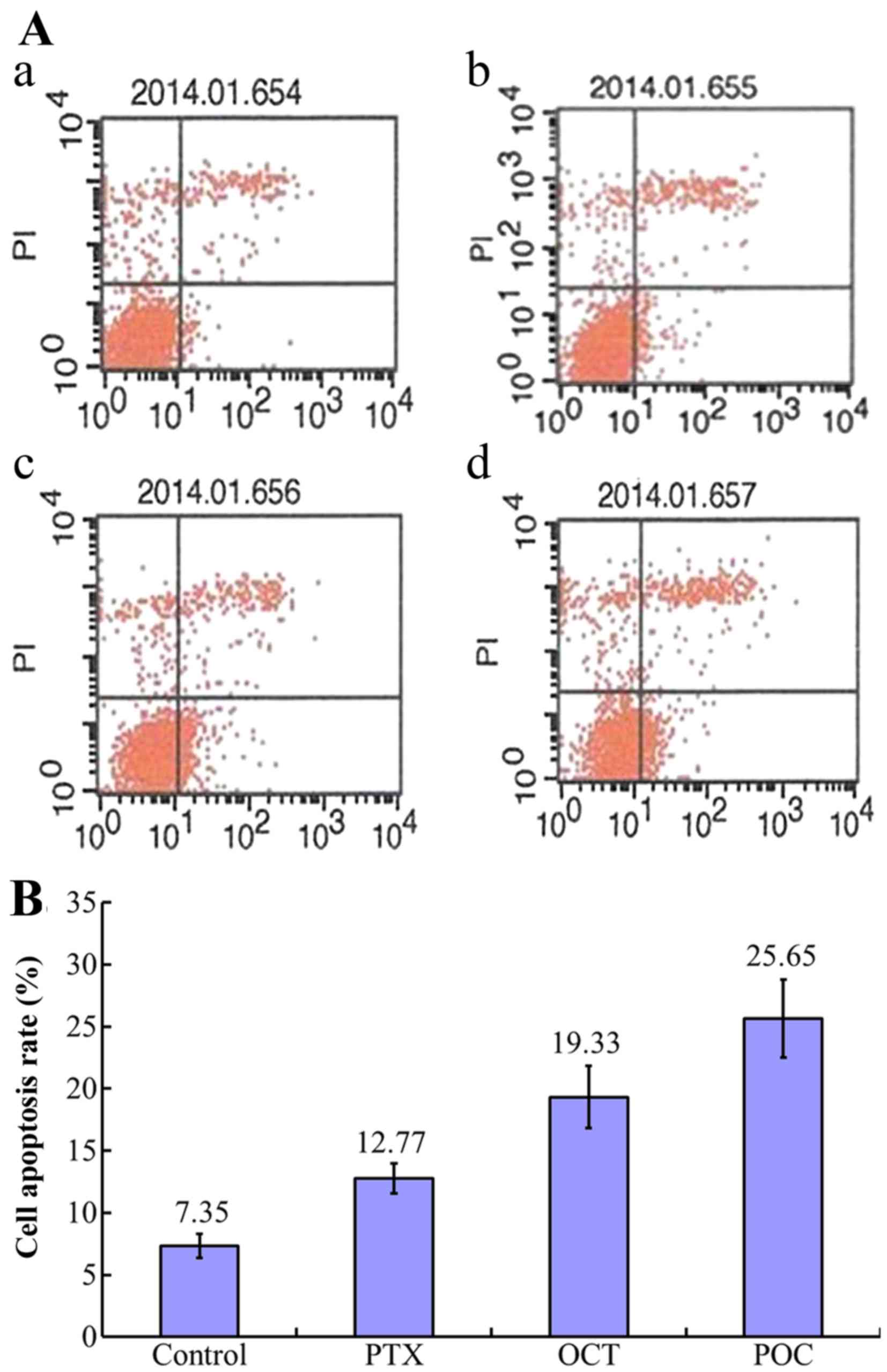
Enhanced apoptotic effect of POC in
A2780/Taxol cells
Compared to the control group, increased apoptosis
was observed in paclitaxel (10 nmol/ml), OCT (10 nmol/ml) and POC
(10 nmol/ml) groups (P<0.05). Both the effect of POC group and
that of OCT group were much more powerful than the paclitaxel group
(P<0.05). In addition, the effect of POC group was more powerful
than the OCT group (P<0.05) (Fig.
6).
Alteration of SSTR2, MDR1 and VEGF
mRNA expression following drug treatment
To investigate the mechanism responsible for the
enhanced effects of POC, mRNA was isolated, and SSTR2,
MDR1 and VEGF mRNA expression was determined
following treatment with POC. As shown in Fig. 7, SSTR2 mRNA was detected in
each group, but there is no difference between the various
concentrations (P>0.05). Compared to the control group, the
expression of both MDR1 and VEGF mRNA decreased in a
dose-dependent manner following 48 h of treatment with POC
(P<0.05), indicating their involvement in the POC-mediated cell
effects.
Alterations of SSTR2, MDR1 protein
expression in A2780/Taxol cells
Western blotting was performed to determine how POC
may affect the protein levels of SSTR2, MDR1 and VEGF. The
intensity of protein bands was read by densitometry and the
standardized results with β-tubulin are summarized in Table III. The results indicated that
following 48 h of treatment, both proteins were decreased in a
dose-dependent manner (P<0.05) (Fig.
8).
| Table III.Effect of POC on expression of SSTR2,
MDR1 and VEGF protein in A2780/Taxol cells. |
Table III.
Effect of POC on expression of SSTR2,
MDR1 and VEGF protein in A2780/Taxol cells.
| Protein | Control | 2.5 nmol/ml | 5.0 nmol/ml | 10.0 nmol/ml |
|---|
| SSTR2 | 1 | 0.486
0.047a | 0.348
0.038a,b | 0.221
0.033a–c |
| MDR1 | 1 | 0.639
0.062a | 0.367
0.057a,b | 0.242
0.027a–c |
| VEGF | 1 | 0.771
0.055a | 0.568
0.035a,b | 0.335
0.030a–c |
| β-tubulin | 1 | 1 | 1 | 1 |
Discussion
Feasibility and simplicity are key factors to be
considered for the design of synthesis strategy. Huang et al
(26) firstly reported that
paclitaxel succinyl anhydride reacts with OCT from solid phase
synthesis through the catalysis of
Benzotriazol-1-yl-oxytripyrrolidino-phosphonium hexafluorophosphate
(PyBOP) and POC is obtained after dissociating from resin. However,
OCT resin complex in this method is not available in the market and
the catalyst PyBOP is expensive which bring us difficulty in
synthesis and application of POC. In this study, we adopted a new
synthesis approach, which overcomes the difficulty of unavailable
OCT resin complex and expensive catalyst. The process included the
sequential synthesis of N-hydroxysuccinimido diphenyl phosphate,
paclitaxel succinyl anhydride, N-hydroxy succinyl acid ester and
POC. Using this method, POC can be synthesized in a large amount
from cheap reagent Diphenyl N-succinimid-ster (SDPP). This has
opened a new route for mass production by pharmaceutical industry,
and thus, opening the door for large scale clinical trials on this
bioactive agent.
Paclitaxel combined with platinum remains the first
line chemotherapy in the treatment of ovarian cancer. However, due
to resistance, it often fails to cure patients. Therefore, the
reversal of paclitaxel resistance in ovarian cancer and increased
sensitivity to paclitaxel-based chemotherapy drugs is a crucial
issue. Our earlier study showed that chemotherapy agent combined
with OCT could markedly inhibit the proliferation and promote
apoptosis of resistant ovarian cancer cells. This combination of
the two single drugs has significant synergistic action (22,23).
Furthermore, studies on synthesized POC in lung cancer cells have
shown its cytotoxicity in vitro and in vivo (26–28).
The thought for increased efficacy of OCT-conjugated taxol was
based on the hypothesis that after binding to SSTR, through SSTR
endocytosis, OCT-conjugated taxol would internalize into the
cytosol of SSTR-expressing tumor cells and therefore increasing the
intracellular concentration of paclitaxel. The present study of
confocal microscopy also supported this opinion. In addition, this
may lead to a decreased toxicity of taxol in non-SSTR-expressing
cells (26).
Our current data suggested that POC inhibited
A2780/Taxol cell proliferation, increased the chemotherapeutic
sensitivity of paclitaxel and reversed chemotherapy resistance.
Moreover, we found that the mRNA levels of SSTR2 were not
altered but the mRNA levels of MDR1 and VEGF were
significantly reduced following POC treatment, whereas, the
expression of SSTR2, MDR1 and VEGF protein appeared to be decreased
by conjugate treatment. The relatively stable levels of SSTR2 mRNA
and significant reduction in protein indicated its
post-translational regulation. Interestingly, Huang et al
(26) reported that short-term OCT
treatment could lead to SSTR2 desensitization, resulting in a
reduced inhibitory effect on hepatocellular carcinoma cells by OCT.
However, long-term OCT treatment effectively inhibited the
development and growth of hepatocellular carcinoma cells probably
via resensitization and upregulation of SSTR2. This discrepancy for
short-term effect on SSTR2 protein levels may be due to the
different characteristics of hepatocellular carcinoma cells and
ovarian cancer cells, the different expression levels of the
receptor, and the higher intracellular concentration that the
conjugate may potentially achieve. Nevertheless, our results have
pointed to the possibility that POC may exert its effect through
decreasing SSTR2 expression.
MDR1 was found to decrease the intracellular
paclitaxel concentration, leading to a reduced or loss of drug
function in ovarian cancer cells (30,31).
This study demonstrated a decreased MDR1 expression in A2780/Taxol
cells by the treatment with POC on both mRNA and protein levels,
suggesting that MDR1 may be involved in POC-mediated inhibition of
cell proliferation and reversal of paclitaxel resistance. Vascular
endothelial growth factor-A (VEGF-A, commonly known as VEGF) is a
key pro-angiogenic factor that plays a crucial role in tumor
expansion (32). Akiyama et
al (33) observed that VEGF
secreted from tumors upregulated MDR1 through the activation of
VEGFR2 and Akt. MDR1 upregulation, via the VEGF-VEGFR pathway in
the tumor microenvironment, is one of the mechanisms of drug
resistance acquired by tumor endothelial cells. This study
demonstrated a decrease of VEGF mRNA and protein expression in
A2780/Taxol cells by POC, suggesting that one action of this agent
may be through the downregulation of MDR1 and inhibition of VEGF
expression. More detailed studies are required to elucidate how the
POC could inhibit the expression of VEGF and the downstream
genes.
The present study demonstrates the effects of POC on
A2780/Taxol cells and explored the possible mechanism mediated by
SSTR2, MDR1 and VEGF. The study introduces a potential
chemotherapeutic reagent for ovarian cancer therapy. However, the
mechanism of the metabolism, transportation and pharmacological
dynamics of POC from the extracellular to intracellular departments
remain unclear. Future mechanistic and in vivo studies are
required for a better understanding of this novel agent.
Acknowledgements
The present study was supported by the Youth
Projects of Jiangsu Provincial Health Department (no. Q201305), the
Science and Technology Research Project of Nanjing City (no.
201201054), a Pre-research Project for National Natural Science
Foundation of Southeast University (no. KJ2010493), and the
Scientific Research Project of Southeast University (no.
3290003101).
References
|
1
|
Syrios J, Banerjee S and Kaye SB: Advanced
epithelial ovarian cancer: From standard chemotherapy to promising
molecular pathway targets - where are we now? Anticancer Res.
34:2069–2077. 2014.PubMed/NCBI
|
|
2
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: The impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McGuire WP, Hoskins WJ, Brady MF, Kucera
PR, Partridge EE, Look KY, Clarke-Pearson DL and Davidson M:
Cyclophosphamide and cisplatin compared with paclitaxel and
cisplatin in patients with stage III and stage IV ovarian cancer. N
Engl J Med. 334:1–6. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sherman-Baust CA, Becker KG, Iii WH Wood,
Zhang Y and Morin PJ: Gene expression and pathway analysis of
ovarian cancer cells selected for resistance to cisplatin,
paclitaxel, or doxorubicin. J Ovarian Res. 4:212011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Szakács G, Paterson JK, Ludwig JA,
Booth-Genthe C and Gottesman MM: Targeting multidrug resistance in
cancer. Nat Rev Drug Discov. 5:219–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gottesman MM, Fojo T and Bates SE:
Multidrug resistance in cancer: Role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Markman M: Combination versus sequential
cytotoxic chemotherapy in recurrent ovarian cancer: Time for an
evidence-based comparison. Gynecol Oncol. 118:6–7. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Theodoropoulou M and Stalla GK:
Somatostatin receptors: From signaling to clinical practice. Front
Neuroendocrinol. 34:228–252. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Watt HL, Kharmate G and Kumar U: Biology
of somatostatin in breast cancer. Mol Cell Endocrinol. 286:251–261.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yano T, Radulovic S, Osuga Y, Kugu K,
Yoshikawa H, Taketani Y and Schally AV: Inhibition of human
epithelial ovarian cancer cell growth in vitro by somatostatin
analog RC-160. Oncology 59 (Suppl 1). 45–49. 2000.
|
|
11
|
Pyronnet S, Bousquet C, Najib S, Azar R,
Laklai H and Susini C: Antitumor effects of somatostatin. Mol Cell
Endocrinol. 286:230–237. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Robbins RJ: Somatostatin and cancer.
Metabolism 45 (Suppl 1). 98–100. 1996.
|
|
13
|
Susini C and Buscail L: Rationale for the
use of somatostatin analogs as antitumor agents. Ann Oncol.
17:1733–1742. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Oberg K: Cancer: Antitumor effects of
octreotide LAR, a somatostatin analog. Nat Rev Endocrinol.
6:188–189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Halmos G, Sun B, Schally AV, Hebert F and
Nagy A: Human ovarian cancers express somatostatin receptors. J
Clin Endocrinol Metab. 85:3509–3512. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jones RH, Reubi JC, Millan D and Vasey P:
Octreotide: An active agent in epithelial ovarian carcinoma? Lancet
Oncol. 5:251–253. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Barnett P: Somatostatin and somatostatin
receptor physiology. Endocrine. 20:255–264. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Annaratone L, Volante M, Asioli S, Rangel
N and Bussolati G: Characterization of neuroendocrine tumors of the
pancreas by real-time quantitative polymerase chain reaction. A
methodological approach. Endocr Pathol. 24:83–91. 2013. View Article : Google Scholar
|
|
19
|
Pawlikowski M: The incidence of
somatostatin receptors in human neoplasms in the light of ex
vivo-in vitro studies. Endokrynol Pol. 57:238–243. 2006.(in
Polish). PubMed/NCBI
|
|
20
|
Murphy E, Prommer EE, Mihalyo M and
Wilcock A: Octreotide. J Pain Symptom Manage. 40:142–148. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hua YP, Yin XY, Peng BG, Li SQ, Lai JM,
Liang HZ and Liang LJ: Mechanisms and influence of
octreotide-induced regulation of somatostatin receptor 2 on
hepatocellular carcinoma. Chemotherapy. 55:312–320. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen Y, Ren M, Shi Y, Zhang Y and Cai Y:
Octreotide enhances the sensitivity of the SKOV3/DDP ovarian cancer
cell line to cisplatin chemotherapy in vitro. Exp Ther Med.
2:1171–1176. 2011.PubMed/NCBI
|
|
23
|
Shen Y, Ren ML, Shi YH, Zhang YX and Cai
YL: Octreotide is the novel alternative for chemosensitivity
enhancement of ovarian cancer cells SKOV3/DDP to cisplatin in vitro
and in nude mice in vivo. Eur J Gynaecol Oncol. 33:584–590.
2012.PubMed/NCBI
|
|
24
|
Chu JJ, Chen KD, Lin YL, Fei CY, Chiang
AS, Chiang CD and Lai YK: Taxol induces concomitant
hyperphosphorylation and reorganization of vimentin intermediate
filaments in 9L rat brain tumor cells. J Cell Biochem. 68:472–483.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Horwitz SB: Mechanism of action of taxol.
Trends Pharmacol Sci. 13:134–136. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang CM, Wu YT and Chen ST: Targeting
delivery of paclitaxel into tumor cells via somatostatin receptor
endocytosis. Chem Biol. 7:453–461. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun ML, Wei JM, Wang XW, Li L, Wang P, Li
M and Yi CH: Paclitaxel-octreotide conjugates inhibit growth of
human non-small cell lung cancer cells in vitro. Exp Oncol.
29:186–191. 2007.PubMed/NCBI
|
|
28
|
Shen H, Hu D, Du J, Wang X, Liu Y, Wang Y,
Wei JM, Ma D, Wang P and Li L: Paclitaxel-octreotide conjugates in
tumor growth inhibition of A549 human non-small cell lung cancer
xenografted into nude mice. Eur J Pharmacol. 601:23–29. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shen Y, Ren ML, Feng X, Cai YL, Gao YX and
Xu Q: An evidence in vitro for the influence of bisphenol A on
uterine leiomyoma. Eur J Obstet Gynecol Reprod Biol. 178:80–83.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lukyanova NY: Characteristics of
homocysteine-induced multidrug resistance of human MCF-7 breast
cancer cells and human A2780 ovarian cancer cells. Exp Oncol.
32:10–14. 2010.PubMed/NCBI
|
|
31
|
Schöndorf T, Kurbacher CM, Göhring UJ,
Benz C, Becker M, Sartorius J, Kolhagen H, Mallman P and Neumann R:
Induction of MDR1-gene expression by antineoplastic agents in
ovarian cancer cell lines. Anticancer Res. 22:2199–2203.
2002.PubMed/NCBI
|
|
32
|
Ebos JM and Kerbel RS: Antiangiogenic
therapy: Impact on invasion, disease progression, and metastasis.
Nat Rev Clin Oncol. 8:210–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Akiyama K, Ohga N, Hida Y, Kawamoto T,
Sadamoto Y, Ishikawa S, Maishi N, Akino T, Kondoh M, Matsuda A, et
al: Tumor endothelial cells acquire drug resistance by MDR1
up-regulation via VEGF signaling in tumor microenvironment. Am J
Pathol. 180:1283–1293. 2012. View Article : Google Scholar : PubMed/NCBI
|