Introduction
Pancreatic cancer is the fourth leading cause of
cancer-related deaths in Japan (1)
and the USA (2). This cancer is
highly resistant to systemic therapies and this is partially the
reason for its high mortality rate (3,4).
Gemcitabine chemotherapy has been the standard treatment for
pancreatic cancer for about a decade; however, gemcitabine plus
nanoparticle albumin-bound (nab)-paclitaxel and FOLFIRINOX
(5-fluorouracil, oxaliplatin, irinotecan and leucovorin) were found
to result in better overall survival (OS) and is now commonly used
(5,6). The drawback however is that these
regimens are more toxic than gemcitabine monotherapy. Moreover,
despite the improvements in chemotherapy, OS is still less than one
year. Therefore, more effective and better-tolerated treatment
options are required to improve the outcome for patients with
advanced pancreatic cancer.
Exciting progress has been made in cancer
immunotherapy with breakthrough immune checkpoint inhibitors and
new cell therapies that enhance the effectiveness of T cells
(7). Programmed death-1 (PD-1)
found on activated T cells is a member of the CD28 family, and is a
key immune checkpoint molecule. When PD-1 binds to programmed
death-ligand 1 (PD-L1) or PD-L2, the T cell receives an inhibitory
signal and no longer mounts productive immune responses (8,9).
Nivolumab, a fully human IgG4 PD-1 antibody, restores anticancer
immune responses by abrogating PD-1 pathway-mediated T cell
inhibition and has been approved for use in Japan and the USA for
treating patients with unresectable melanoma (9,10). The
clinical efficacy of nivolumab has also been reported in patients
with non-small cell lung cancer (11) and renal cell carcinoma (12). In addition to PD-1 and PD-L1, other
immune checkpoint inhibitors are currently being developed for
various tumors (13,14).
PD-1 or PD-L1 antibodies have been shown to exert a
substantial antitumor effect in mouse pancreatic cancer models
(15,16). Although no studies to date have
demonstrated the clinical efficacy of immune checkpoint inhibitor
monotherapy for pancreatic cancer (17,18),
ipilimumab (a checkpoint inhibitor of CTLA-4) combined with cancer
vaccine has shown a survival benefit (19,20).
Thus, immune checkpoint inhibitors combined with immune- or
non-immune-based therapies are great prospective treatment
strategies for pancreatic cancer.
It is still unclear what impact anticancer agents
have on immune checkpoint molecules when chemotherapy is combined
with immunotherapy. Very few studies have addressed the PD-1/PD-L1
pathways, and there are contradictions among them (21–24).
It is within this context that we investigated how anticancer
agents influence PD-L1 expression in pancreatic cancer cell lines.
In the present study, we demonstrated that commonly used anticancer
agents for pancreatic cancer (i.e. gemcitabine, 5-fluorouracil and
paclitaxel) upregulate cell surface PD-L1 expression in both human
and mouse pancreatic cancer cell lines. Additionally, we provide
evidence that not only the MAPK and PI3K/AKT pathway, which are
known pathways, but also the JAK/STAT pathway is involved in the
induction of PD-L1 expression in response to these anticancer
agents.
Materials and methods
Cell lines and reagents
The human pancreatic cancer cell line MIA PaCa-2 was
obtained from Riken BioResource Center (Tsukuba, Japan) and AsPC-1
cells were obtained from the American Type Culture Collection
(ATCC; Rockville, MD, USA). The murine pancreatic cancer cell line
Pan02, which is syngeneic to C57Bl/6 mice, was purchased from the
Division of Cancer Treatment and Diagnosis, National Cancer
Institute (Bethesda, MD, USA). AsPC-1 and Pan02 cells were grown in
75 cm2 cell culture flasks and maintained in Roswell
Park Memorial Institute (RPMI)-1640 medium supplemented with 10%
fetal bovine serum (FBS), L-glutamine and penicillin (100
U/ml)/streptomycin (100 µg/ml) (both from Gibco Life Technologies,
Carlsbad, CA, USA). MIA PaCa-2 cells were cultured in Dulbecco's
modified Eagles medium low glucose supplemented with 10% FBS,
L-glutamine and penicillin (100 U/ml)/streptomycin (2.5 µg/ml).
Cells were incubated at 37̊C in a humidified atmosphere containing
5% CO2.
In the present study, we used gemcitabine (GEM),
5-fluorouracil (5-FU) and paclitaxel (PTX) which are agents
commonly used to treat pancreatic cancer; all agents were
immediately prepared before use. GEM was purchased from the Tokyo
Chemical Industry Co., Ltd. (Tokyo, Japan), 5-FU from Kyowa Hakko
Kirin Co., Ltd. (Tokyo, Japan) and PTX was obtained from Nippon
Kayaku Co., Ltd. (Tokyo, Japan).
Evaluation of PD-L1 expression after
exposure to anticancer agents
Cells were seeded at 3.0–4.5×105
cells/well in 6-well plates 48 h before treatment and left to
incubate at 37̊C in a humidified atmosphere containing 5%
CO2. After 48 h, cells were exposed to various
concentrations of GEM, 5-FU and PTX for 6–72 h. For each drug, the
concentration used in our experiment was based on their plasma
level in clinical use. The expression level of PD-L1 was determined
using flow cytometry and qRT-PCR.
Treatment with JAK/STAT and other
pathway inhibitors
AsPC-1 cells were adjusted to 4.5×105
cells/well into 6-well plates. After a 48-h incubation at 37̊C in a
humidified atmosphere containing 5% CO2, the cells were
treated for 1 h with various concentrations of JAK2 (AG490; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA), Akt (perifosine) or
MEK1/2 inhibitors (U012) (both from Cell Signaling Technology,
Inc., Danvers, MA, USA). Cells were stimulated with 5-FU, PTX and
GEM and then incubated in the presence or absence of AG490 for an
additional 6–48 h. PD-L1 expression was analyzed using flow
cytometry and qRT-PCR.
Western blotting
Cells were washed twice with phosphate-buffered
saline (PBS) and lysed in ice-cold lysis buffer (CelLytic™ MT cell
lysis reagent) containing 2% proteinase inhibitor (both from
Sigma-Aldrich Co., St. Louis, MO, USA). Cells were retrieved with a
cell scraper, stirred and incubated on ice for 15 min. Lysates were
centrifuged, supernatants were collected, and protein concentration
was determined using the Bio-Rad protein assay (Bio-Rad
Laboratories, Hercules, CA, USA). The supernatants were diluted
with NuPAGE LDS sample buffer (Life Technologies, Grand Island, NY,
USA) to create equal concentrations of protein. Ten micrograms of
protein were separated on 4–12% NuPage Bis-Tris gels and blotted
onto a nitrocellulose membrane using the iBlot Dry Blotting System
(all from Life Technologies) according to the manufacturer's
protocol. Blots were blocked with 10% EzBlock Chemi (ATTO
Corporation, Tokyo, Japan) in TBS-T [10 mM Tris-HCl (pH 8.0), 150
mM NaCl, 0.1% Tween-20 v/v] for 1 h at room temperature and washed
with TBS-T 3 times. The membranes were incubated overnight at 4̊C
with anti-STAT1, anti-phospho-STAT1, anti-NF-κB (all from Cell
Signaling Technology, Inc.), anti-Akt1/2/3, anti-phospho-Akt1/2/3,
anti-p38, anti-phospho-p38 (all from Santa Cruz Biotechnology,
Inc.), anti-JNK (Cell Signaling Technology, Inc.), anti-phospho-JNK
(Santa Cruz Biotechnology, Inc.), anti-Erk 1/2 (Cell Signaling
Technology, Inc.), anti-phospho-Erk 1/2, anti-tubulin (both from
Santa Cruz Biotechnology, Inc.) and anti-β actin (Abcam, Cambridge,
MA, USA) antibodies in TBS-T (diluted 1:1,000). After washing in
TBS-T, the membranes were incubated with the secondary anti-rabbit
and mouse IgG antibodies (Life Technologies) in TBS-T (diluted
1:10,000) for 1 h at room temperature. Immunocomplexes were
detected using western blotting (ECL Prime; GE Healthcare UK Ltd.,
Buckinghamshire, UK).
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
The expression level of PD-L1 was determined using
real-time PCR as previously described (25). The samples used for mRNA isolation
were removed from the pancreatic cancer cells (AsPC-1, MIA PaCa-2
and Pan02). Total mRNA was extracted using the acid guanidinium
phenol chloroform method with Isogen (Nippon Gen Co. Ltd., Tokyo,
Japan). The isolated RNA was stored at −80°C until use for
real-time PCR. In the latter, 1 µg of extracted RNA was
reverse-transcribed. The resulting cDNA was subjected to qRT-PCR
using the following primers for human PD-L1: (forward primer,
5′-GTACCGCTGCATGATCAGCTAT-3′ and reverse primer,
5′-GGCATTGACTTTCACAGTAATTCG-3′); murine PD-L1 (forward primer,
5′-CAGGCCGAGGGTTATCCA-3′ and reverse primer,
5′-CGGGTTGGTGGTCACTGTTT-3′); human GAPDH (forward primer,
5′-ACCACAGTCCATGCCATCACT-3′ and reverse primer, CCATCACGCCACAGTTT
CC); and murine β-actin (forward primer,
5′-TATCCACCTTCCAGCAGATGT-3′ and reverse primer,
5′-AGCTCAGTAACAGTCCGCCTA-3′). PCR was performed using a Power
SYBR-Green PCR Master Mix and a real-time PCR system (7300; Applied
Biosystems, Foster City, CA, USA). Relative quantifications of gene
expression with qRT-PCR data were calculated relative to human
GAPDH or murine β-actin.
Flow cytometric analysis
PD-L1 surface expression was analyzed by flow
cytometry. Cells harvested from in vitro cultures were
washed twice with CellWash™ (Becton-Dickinson and Co., Franklin
Lakes, NJ, USA) and then incubated with anti-PD-L1 (human,
Becton-Dickinson and Co., mouse, eBioscience, San Diego, CA, USA);
or isotype control antibodies (eBioscience) for 30 min at 4̊C. The
cells were washed with CellWash once and analyzed by flow cytometer
on a FACSCalibur flow cytometer and CellQuest™ Pro version 6.0
software (both from Becton-Dickinson and Co.).
Statistical analysis
Results are expressed as means ± standard error of
the mean (SEM). Comparisons among groups or against one control
group were evaluated by one-way ANOVA test followed by the
Tukey-Kramer's or Dunnett's post hoc multiple comparisons test,
respectively. Statistical significance was taken as P<0.05.
Statistical analyses were performed with EZR (Saitama Medical
Center, Jichi Medical University, Saitama, Japan), which is a
graphical user interface for R (The R Foundation for Statistical
Computing, Vienna, Austria). More precisely, it is a modified
version of R commander designed to add statistical functions
frequently used in biostatistics (26).
Results
Anticancer agents upregulate PD-L1
surface expression in pancreatic cancer cell lines
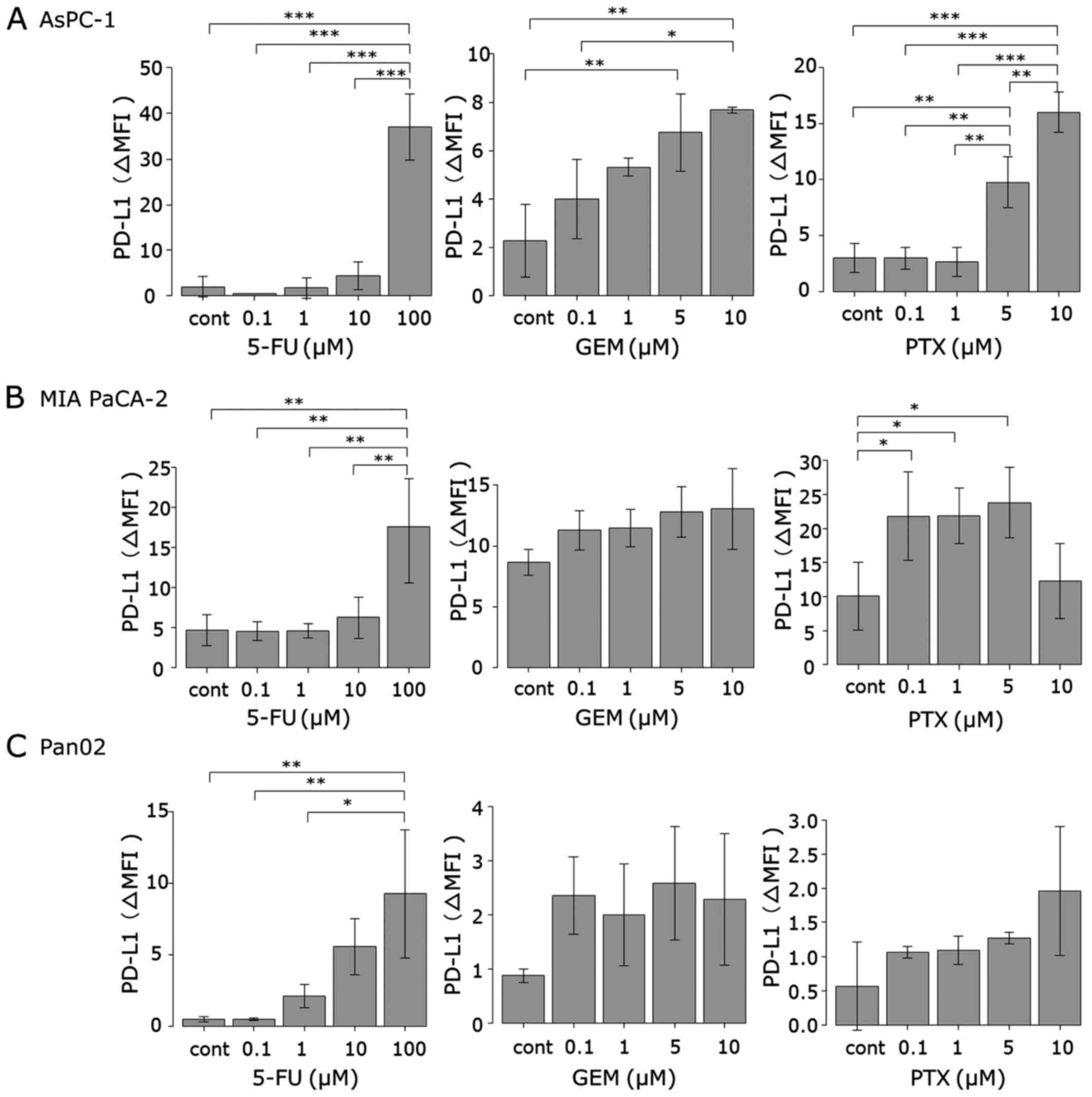
MIA PaCa-2, AsPC-1 and Pan02 cells were treated with
GEM, 5-FU and PTX for 24–72 h to determine whether they can induce
PD-L1 protein expression. Expression was determined using flow
cytometry and is expressed as the Δ mean fluorescence intensity
(ΔMFI; MFI using anti-PD-L1 subtracted from the isotype control).
As shown in Figs. 1 and 2, treatment with 5-FU for 72 h induced
PD-L1 surface expression in both human pancreatic cancer cell lines
(MIA PaCa-2 and AsPC-1) at 100 µM, whereas 5-FU did not affect the
expression at concentrations <100 µM. In mouse pancreatic cancer
cell line Pan02, 5-FU (72 h) at concentrations >1 µM induced
PD-L1 expression in a dose-dependent manner. Treatment with GEM for
24 h induced PD-L1 expression in the AsPC-1 cells in a
dose-dependent manner. In both MIA PaCa-2 and Pan02 cell lines, GEM
induced PD-L1 expression at each concentration, but it did not
reach statistical significance. Treatment with PTX for 48 h
significantly induced PD-L1 expression in the AsPC-1 cells at
concentrations of 5 and 10 µM and in MIA PaCa-2 cells at each
concentration except 10 µM. PTX also induced PD-L1 expression in
mouse Pan02 cells at each concentration, but it did not reach
statistical significance.
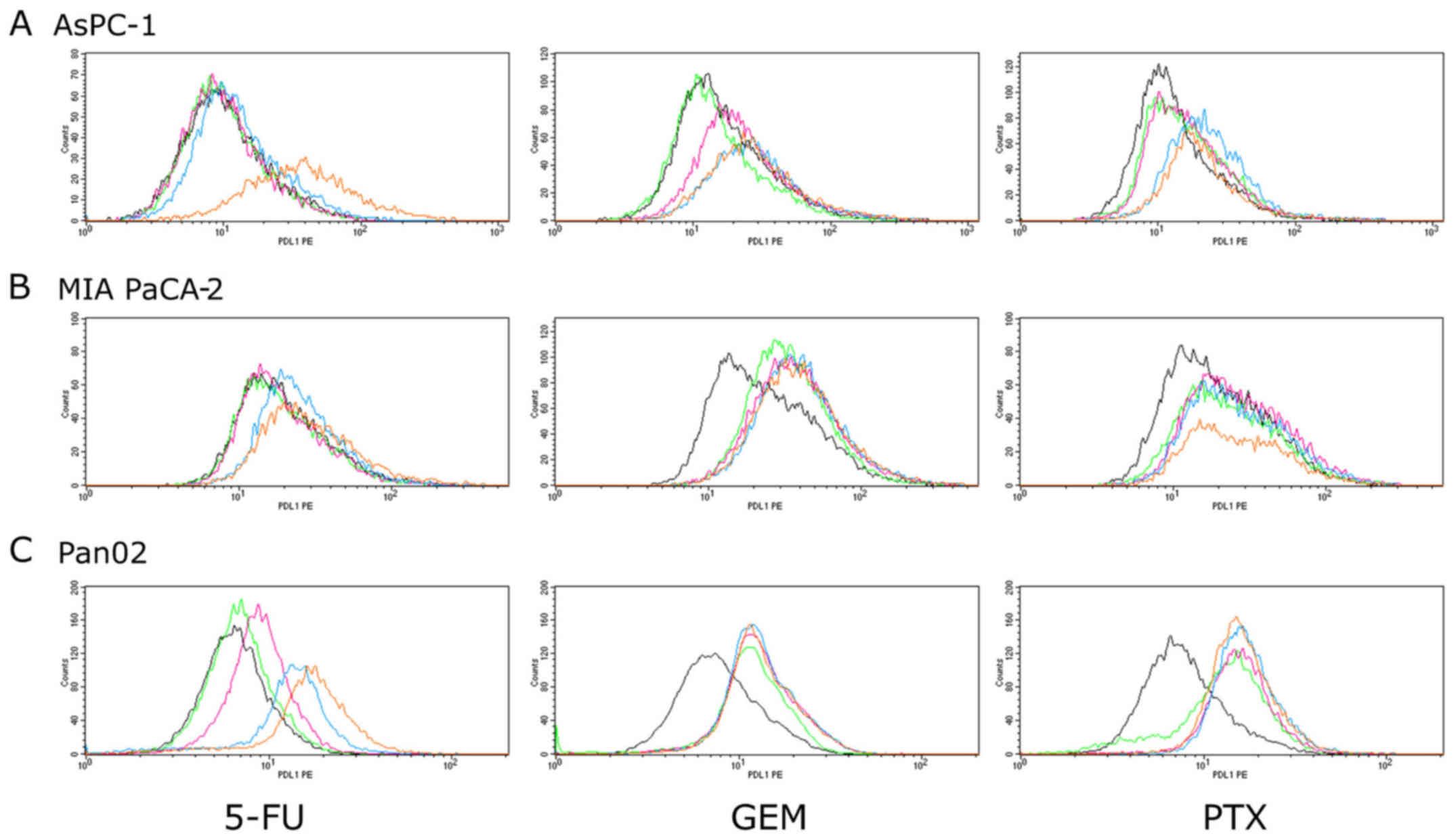 | Figure 2.PD-L1 surface protein expression in
pancreatic cancer cell lines. (A) AsPC-1, (B) MIA PaCa-2 and (C)
Pan02 cells were stimulated by three anticancer agents (5-FU for 72
h, GEM for 24 h and PTX for 48 h). PD-L1 expression was then
analyzed using flow cytometry. The concentrations of each
anticancer agents are indicated as follows: 5-FU: black, 0 µM;
green, 0.1 µM; pink, 1 µM; blue, 10 µM; orange, 100 µM. GEM and
PTX: black, 0 µM; green, 0.1 µM; pink, 1 µM; blue, 5 µM; orange, 10
µM. 5-FU, 5-fluorouracil; GEM, gemcitabine; PTX, paclitaxel. |
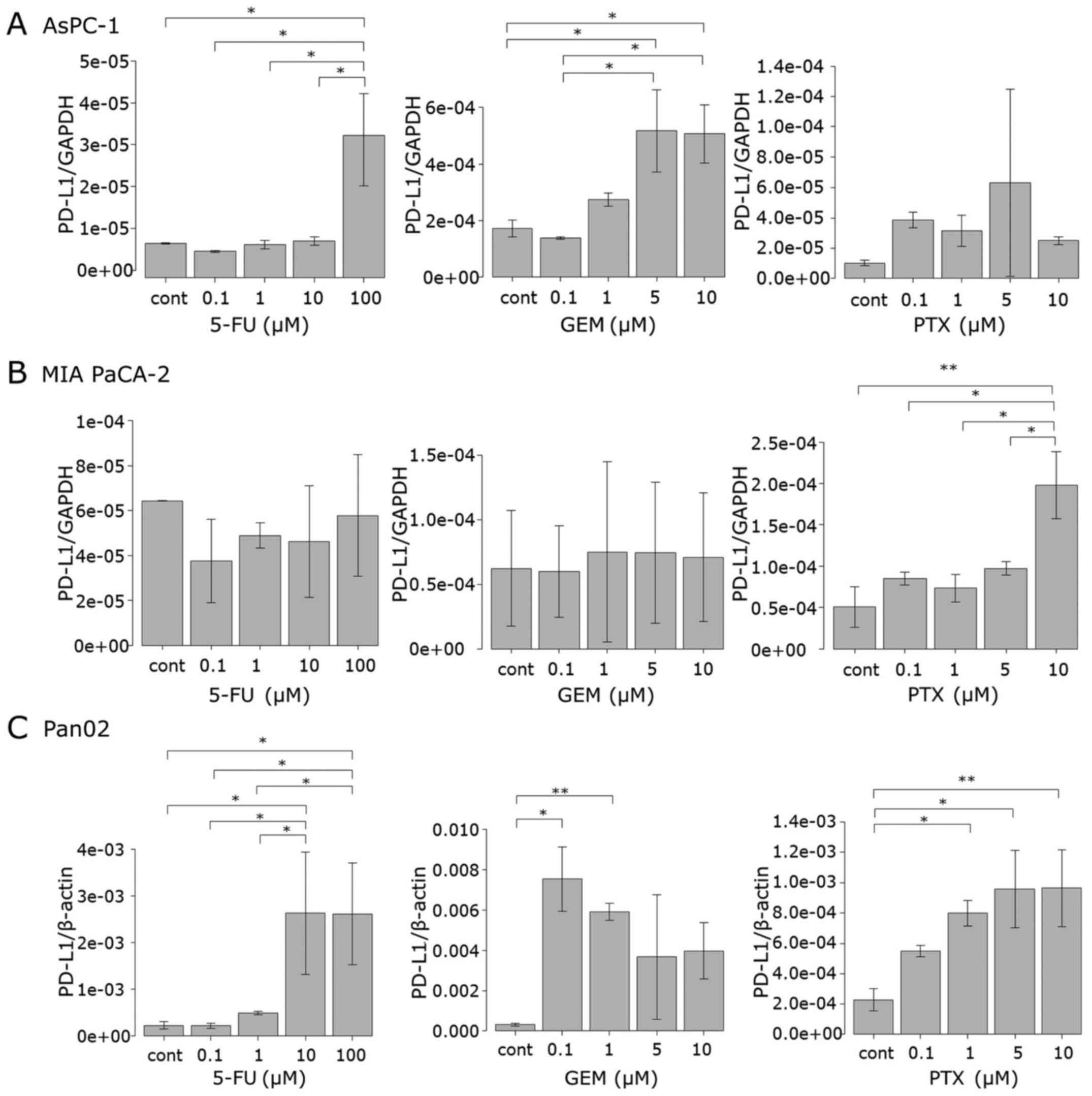
Anticancer agents induce PD-L1 mRNA
expression in pancreatic cancer cell lines
To investigate how the anticancer agents induce
PD-L1 protein expression in tumor cells, the mRNA level of PD-L1
was determined by qRT-PCR. MIA PaCa-2, AsPC-1 and Pan02 cells were
treated with 5-FU, GEM and PTX for 24 h in a manner similar as that
carried out for the protein assessment as described above. In
AsPC-1 cells, 5-FU significantly increased the PD-L1 mRNA level at
100 µM, as observed in the surface protein expression. Treatment
with GEM at 5 and 10 µM significantly upregulated mRNA expression
while each dose of PTX upregulated mRNA expression although not to
statistically significant levels (Fig.
3A). In the MIA PaCa-2 cells, neither 5-FU nor GEM affected
PD-L1 mRNA expression, whereas PTX at 10 µM significantly increased
the mRNA level (Fig. 3B). The PD-L1
mRNA expression pattern in the Pan02 cells was similar to that of
the surface protein, that is, the mRNA level increased when 5-FU
exceeded 1 µM, while GEM and PTX upregulated the mRNA level at each
concentration (Fig. 3C).
Anticancer agents activate the
JAK/STAT pathway
The protein and mRNA expression analyses suggest
that surface PD-L1 protein expression in the AsPC-1 and Pan02 cells
was regulated mainly at the mRNA level. As such, we used the human
AsPC-1 cell line to ascertain the molecular mechanisms by which the
anticancer agents induced PD-L1 mRNA expression. It has been
reported that the JAK/STAT pathway is deeply involved in
IFN-γ-mediated PD-L1 upregulation in solid tumors such as lung
cancer and hepatocellular carcinoma (27,28).
In contrast, this pathway has not been implicated in anticancer
agent-mediated PD-L1 expression. As such, we aimed to ascertain
whether our three anticancer agents activate the JAK/STAT signaling
pathway in AsPC-1 cells. AsPC-1 cells were exposed to 5-FU (100 µM)
or GEM (10 µM) for 48 h, and PTX (10 µM) for 24 h following which
the expression level and phosphorylation of STAT1 were determined
using western blotting. The phosphorylation of STAT1 was greatly
induced by each of the three anticancer agents (Fig. 4). The protein level of STAT1 was
increased after 5-FU and GEM treatment, whereas it remained
unchanged after PTX treatment.
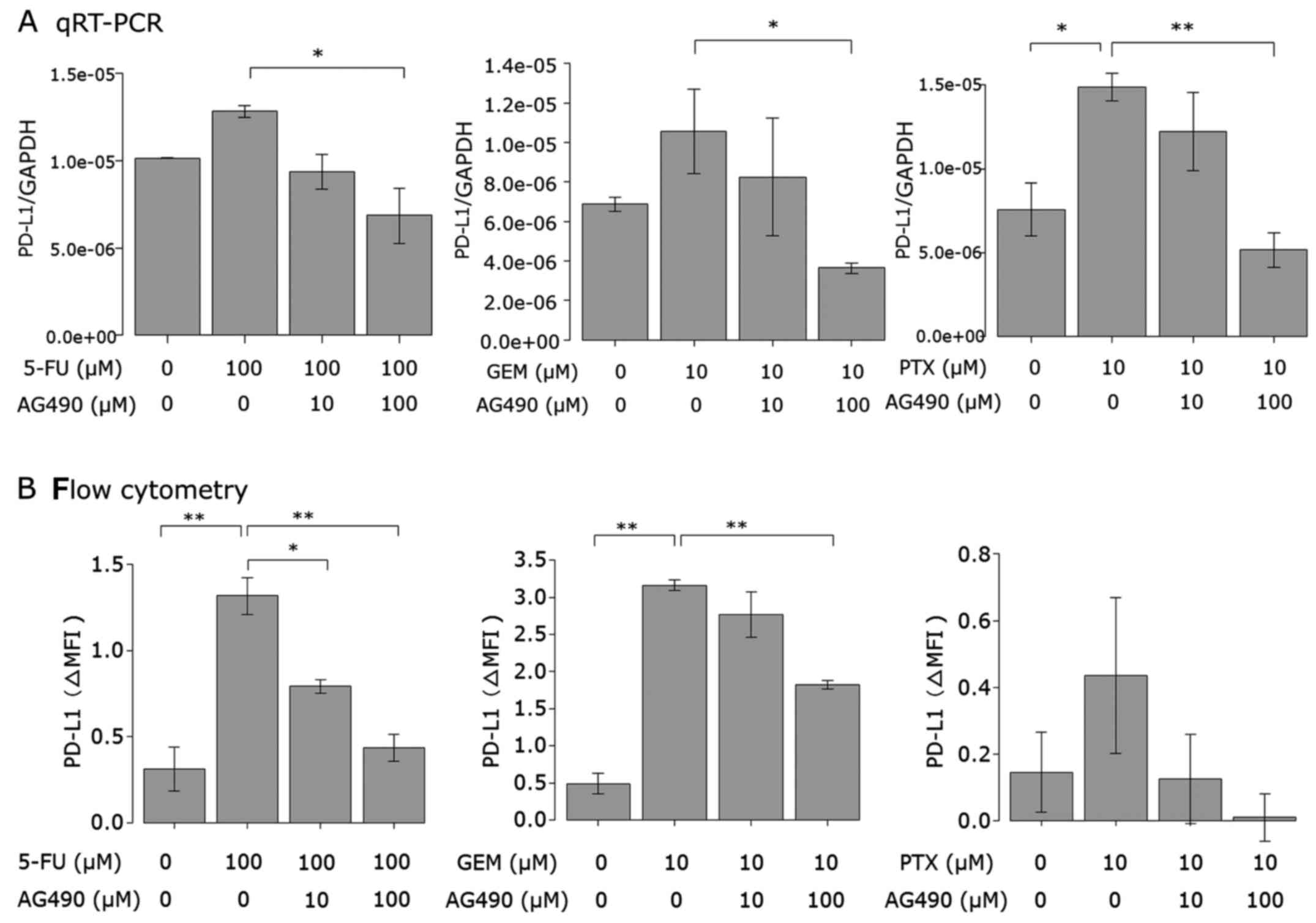
Anticancer agent-induced PD-L1
expression is attenuated by blocking the JAK/STAT pathway
Next, we examined the effect of a JAK2 inhibitor on
the upregulation of PD-L1 expression induced by the anticancer
agents in AsPC-1 cells. Cells were treated with the JAK2 inhibitor
AG490 or left untreated prior to 5-FU, PTX or GEM stimulation. Both
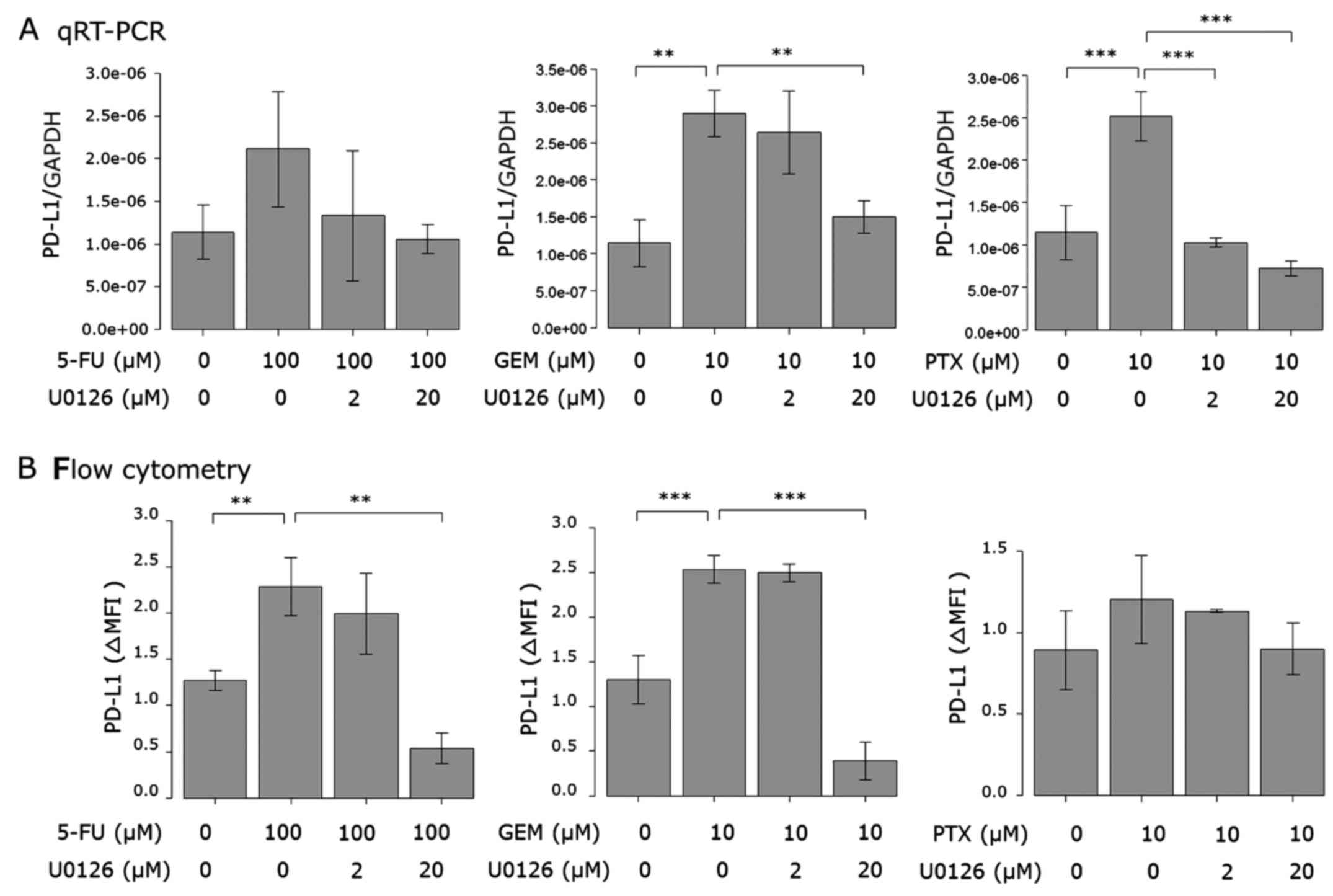
PD-L1 mRNA expression (Fig. 5A) and
cell surface protein (Fig. 5B)
induced by the anticancer agents were attenuated by AG490 in a
dose-dependent manner. AG490 at 100 µM almost completely attenuated
the PD-L1 expression induced by the anticancer agents.
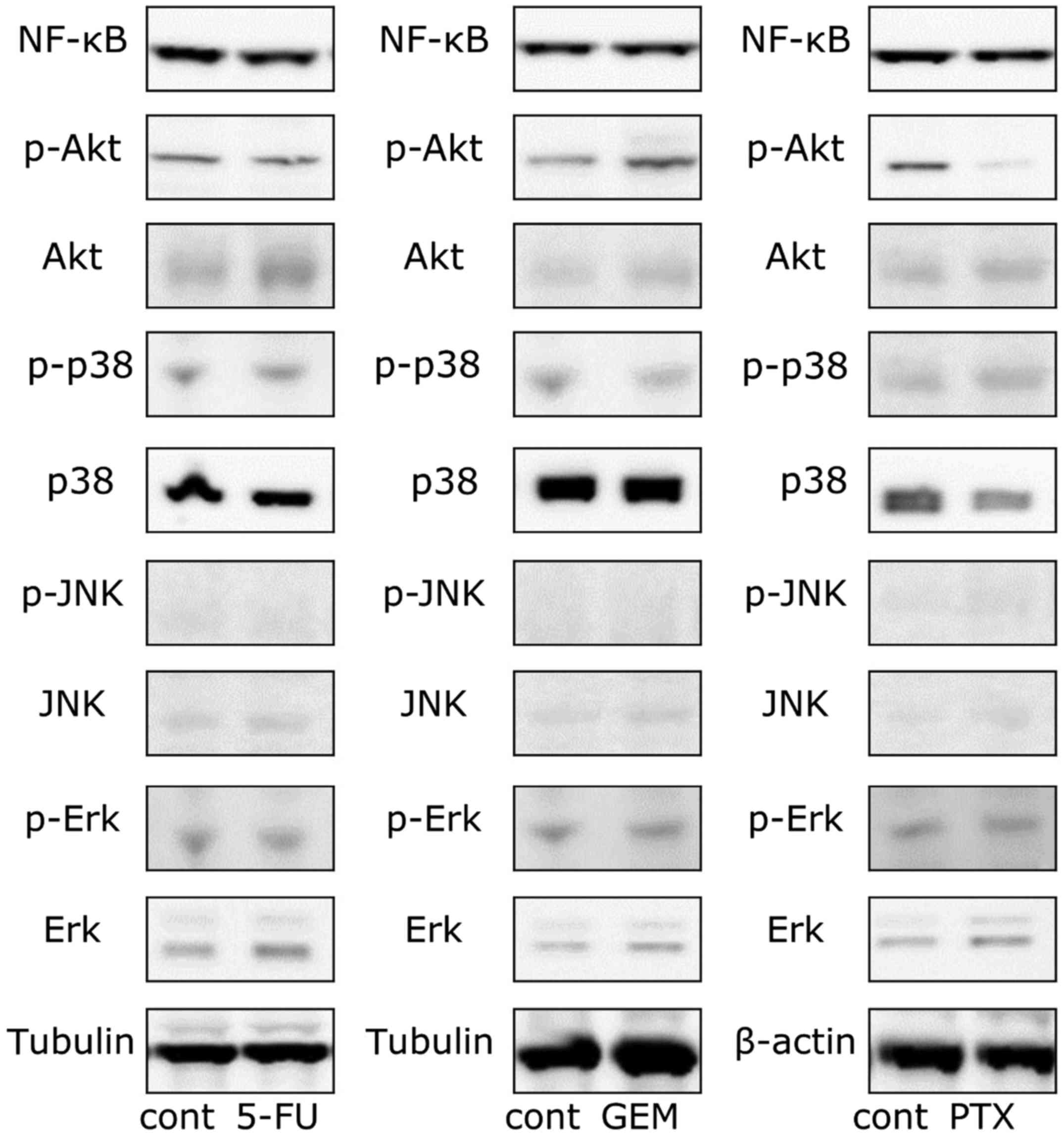
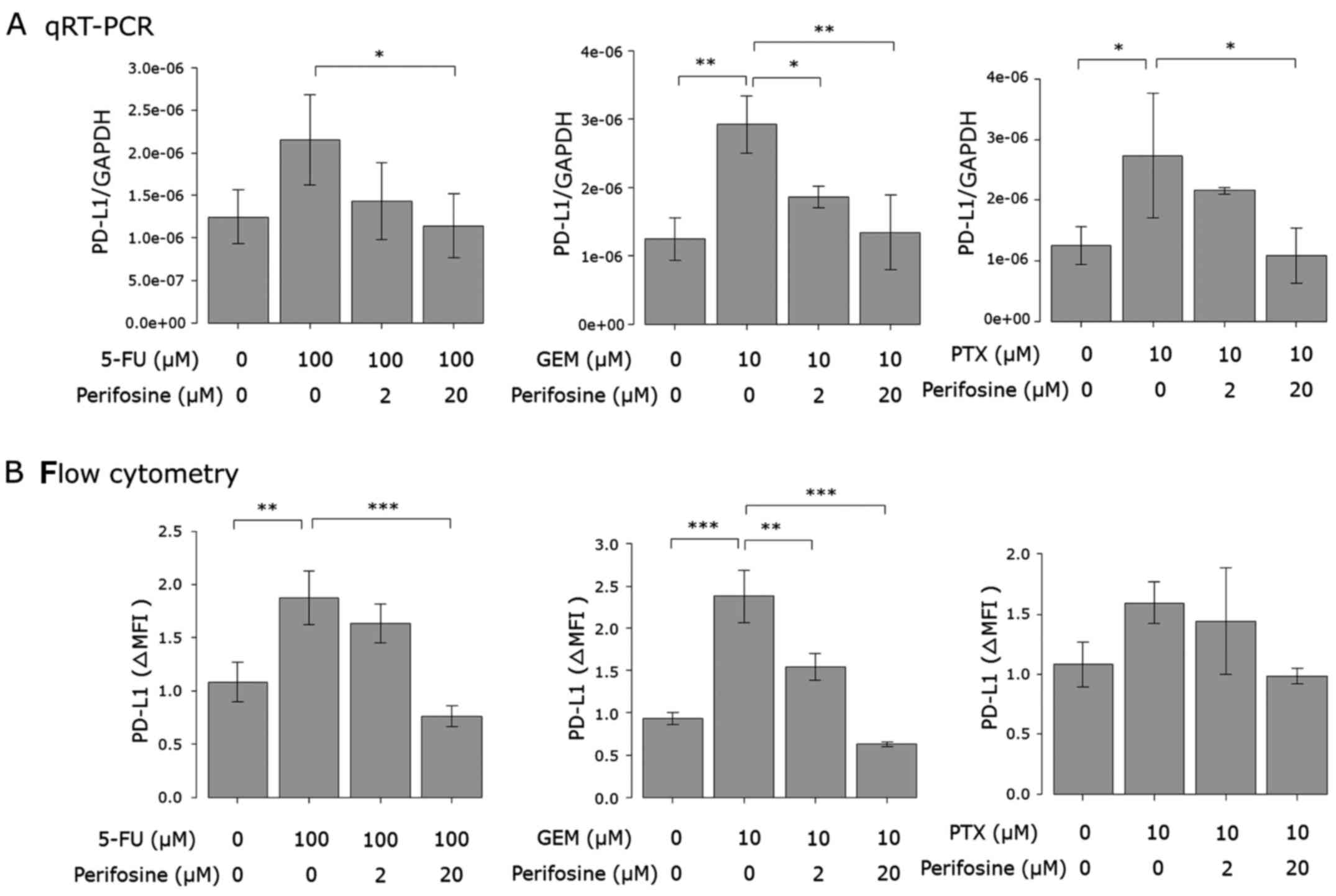
Involvement of other pathways
Additionally, we also investigated whether the
PI3K/AKT (29), NF-κB (24,30)
and MAPK (23,31) pathways respond to anticancer agents,
since they were previously implicated in anticancer agent-induced
PD-L1 expression. The phosphorylation of Akt was induced by only
GEM stimulation, although its protein level was enhanced by 5-FU
and GEM (Fig. 6). The protein level
of Erk was enhanced by each of the three anticancer agents. The
intracellular protein level of NF-κB and the phosphorylation and
protein levels of p38 were not affected by the drugs (Fig. 6). Considering the results of the
western blot analysis, we also investigated the effect of the
inhibitors of the PI3K/AKT and MAPK pathways. As shown in Figs. 7 and 8, anticancer agent-induced PD-L1
upregulation was attenuated by the Akt and MEK1/2 inhibitors.
Discussion
The success of immune checkpoint inhibitors and new
generations of adoptive cell transfer therapy, such as chimeric
antigen receptor (CAR) T cell therapy, have highlighted the
potential of immunotherapy as a treatment option for various tumors
(7,32). Moreover, extensive effort is being
directed toward effectively combining immune- and non-immune-based
therapies. For example, combining immune therapy and anticancer
agents is being explored, albeit the impact of anticancer agents on
the expression of immune checkpoint molecules is not yet fully
understood. In the present study, we present two major findings.
Firstly, PD-L1 surface expression in pancreatic cancer cell lines
was upregulated by 5-fluorouracil, paclitaxel and gemcitabine.
Secondly, the JAK/STAT pathway as well as other known pathways
(i.e. MAPK and PI3K pathway) were involved in this PD-L1
upregulation. To the best of our knowledge, this is the first study
to address the effect of anticancer agents on PD-L1 expression in
pancreatic cancer cells and the involvement of the JAK/STAT pathway
in the anticancer agent-mediated PD-L1 expression.
The effect of chemotherapy agents on PD-L1
expression has been discussed in four studies that we are aware of.
However, there is a lack of consensus and the topic remains
controversial (21–24). Among them, three studies
demonstrated that the anticancer agents upregulated surface PD-L1
expression, while the fourth reported the downregulation of surface
PD-L1. In their study, Zhang et al observed that paclitaxel,
etoposide and 5-fluorouracil induced PD-L1 surface expression in
human breast cancer cell lines (21), however they did not study the
molecular mechanisms that led to the increase in PD-L1 expression.
Gong et al reported that paclitaxel induced PD-L1 surface
protein and mRNA expression in both human colorectal (SW480) and
human hepatocellular carcinoma (HepG2) cell lines (23). They demonstrated that paclitaxel
treatment induced Erk 1/2 phosphorylation in both cell lines and
the increase in PD-L1 expression caused by paclitaxel was partially
blocked by an MEK inhibitor. Peng reported that PD-L1 expression in
ovarian cancer cell lines was augmented via NF-κB signaling by
paclitaxel, gemcitabine or carboplatin treatment (24). In contrast, Ghebeh et al
reported that doxorubicin downregulated the surface expression of
PD-L1 in breast cancer cells and upregulated nuclear expression of
PD-L1 by means of the PI3K/AKT pathway (22). One possible explanation for the
difference among these four studies, as well as our own, may be
attributed to differences in the cell lines and anticancer agents
used in each study. In the present study, we used 5-fluorouracil,
gemcitabine and paclitaxel since they are commonly used alone or
combined with other agents when treating pancreatic cancer. The
concentration of each anticancer agent in our experiments was based
on the plasma level of each drug when clinically used (33,34) or
the concentration that was used in previous in vitro
experiments (21,35,36).
Consequently, the maximum concentration of 5-fluorouracil in our
experiment was 10-fold higher than that used for gemcitabine and
paclitaxel. This difference in drug concentration among the
anticancer agents may have influenced the degree of PD-L1 induction
by the agents.
In the present study, PD-L1 surface protein
expression was enhanced in all pancreatic cancer cell lines. The
absolute value of PD-L1 expression determined by flow cytometry or
qRT-PCR varied with each experiment; this could partly result from
PD-L1 expression being affected by cell conditions. PD-L1
expression was consistently upregulated when stimulated by the
anticancer agents; the pattern of this relative change was
identical in each experiment. We observed that PD-L1 at the mRNA
level was upregulated as well as surface protein expression when
AsPC-1 or Pan02 cells were stimulated by each anticancer agent.
Meanwhile, the mRNA level of PD-L1 in the MIA PaCa-2 cells did not
increase when cells were treated with 5-fluorouracil, gemcitabine
or lower concentrations of paclitaxel. It was reported that
doxorubicin alters PD-L1 surface expression by a
post-transcriptional regulation mechanism that involves the
translocation of the protein from the membrane to the nucleus
(22). Since anticancer agents
induced PD-L1 surface protein expression without upregulating mRNA
in the MIA PaCa-2 cells, it is possible that the expression was
mainly regulated at a post-transcriptional level in this cell
line.
In regards to the mechanism of PD-L1 regulation,
Pardoll reported that innate and adaptive immune resistance are the
two general mechanisms by which tumor cells regulate PD-L1
(37). In innate immune resistance,
PD-L1 expression is driven by constitutive oncogenic signaling
pathways such as the PI3K/AKT pathway (38) and STAT3 (39). In contrast, in adoptive immune
resistance, several signaling pathways such as JAK/STAT (27,28),
PI3K (29), MAPK (29) and NF-κB (29,30)
appear to be involved in PD-L1 expression induced by IFN-γ. Only
the PI3K/AKT (22), MAPK (23) and NF-κB (24) pathways have been previously reported
to be involved in the upregulation of PD-L1 induced by anticancer
agents, but involvement of the JAK/STAT pathway has not yet been
reported. To obtain a better understanding of the mechanisms
involved in PD-L1 upregulation, we sought to determine whether the
JAK/STAT pathway regulates PD-L1 transcription. We found that each
of the three anticancer agents induced the phosphorylation of STAT1
in the AsPC-1 cells, and JAK2 inhibitor AG490 reversed the
upregulation of PD-L1 induced by the anticancer agents at both the
mRNA and protein levels. These findings indicate that the JAK/STAT
pathway regulates the expression of PD-L1. Other previously
reported signaling pathways (i.e. MAPK and PI3K pathways) are also
implicated in the present study. The relative importance of these
pathways is unknown at the present, but we believe that these
pathways are intricately involved in the anticancer agent-mediated
PD-L1 expression by signaling crosstalk.
The discoveries made in the present study are a
critical step towards further uncovering the mechanism of PD-L1
expression in pancreatic cell lines albeit with a few limitations.
In the present study, we examined the signaling pathways in AsPC-1
cells only, therefore it is unclear whether these results apply to
other cell lines. Pancreatic cancer cell lines including AsPC-1 and
MIA PaCa-2 have different genetic alterations such as the KRAS
(v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog), TP53
(encoding the p53 protein) and SMAD4 (SMAD family member 4, also
known as DPC4; deleted in pancreatic carcinoma locus 4) gene
mutations, which affect growth characteristics, tumorigenicity and
chemosensitivity (40). AsPC-1 is
considered more resistant to gemcitabine than MIA PaCa-2 (40,41).
Gemcitabine resistance has been liked to signaling pathways
associated with PD-L1 expression such as JAK/STAT, MAPK, PI3K-AKT
and NF-κB (42–45). These genetic alterations and the
difference in the cell signaling response to anticancer agents can
also alter the PD-L1 expression induced by the anticancer agents.
Future research using additional cell lines is needed to clarify
the relationship between these genotypic differences among cell
lines and their effect on the PD-L1 expression induced by
anticancer agents.
In conclusion, our results indicate that
5-fluorouracil, gemcitabine and paclitaxel enhance PD-L1 expression
in pancreatic cancer cell lines via several pathways including the
JAK/STAT pathway. Pancreatic cancer is still intractable due to its
resistance to conventional treatments including anticancer agents.
Our results imply that anticancer agents not only cause
cytotoxicity, but also alter the tumor immune response which may
induce tumor immune escape. Cancer immunotherapy including blockade
of PD-1/PD-L1 is expected to become the new standard therapy for
many cancers and combination strategy in immunotherapy is currently
being developed. We believe that the data provided in the present
study, may aid in the design of more effective treatments that
combine chemotherapy and immunotherapy.
Acknowledgements
The present study was supported by Grant-in-Aid for
Scientific Research (C) to T.I. (no. 26460914) and Grant-in-Aid for
Young Scientist (B) to T.O. (no. 26830112) from the Ministry of
Education, Culture, Sports, Science and Technology of Japan.
Yoshito Itoh received a lecture fee and is affiliated with a
department that was partially funded by Bristol-Myers Squibb, and
receives a grant from Kyowa Hakko Kirin Co. Ltd.
Glossary
Abbreviations
Abbreviations:
|
PD-1
|
programmed death-1
|
|
PD-L1
|
programmed death-ligand 1
|
|
JAK
|
Janus activated kinase
|
|
STAT
|
signal transducer and activator of
transcription
|
|
GEM
|
gemcitabine
|
|
5-FU
|
5-fluorouracil
|
|
PTX
|
paclitaxel
|
References
|
1
|
Statistics and Information Department,
Ministry of Health, . Labour and Welfare: Vital Statistics. Tokyo:
2013
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mazur PK and Siveke JT: Genetically
engineered mouse models of pancreatic cancer: Unravelling tumour
biology and progressing translational oncology. Gut. 61:1488–1500.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dorado J, Lonardo E, Miranda-Lorenzo I and
Heeschen C: Pancreatic cancer stem cells: New insights and
perspectives. J Gastroenterol. 46:966–973. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al: Groupe Tumeurs Digestives of Unicancer;
PRODIGE Intergroup: FOLFIRINOX versus gemcitabine for metastatic
pancreatic cancer. N Engl J Med. 364:1817–1825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Couzin-Frankel J: Breakthrough of the year
2013. Cancer immunotherapy. Science. 342:1432–1433. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen L: Co-inhibitory molecules of the
B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol.
4:336–347. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brahmer JR, Hammers H and Lipson EJ:
Nivolumab: Targeting PD-1 to bolster antitumor immunity. Future
Oncol. 11:1307–1326. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Larkin J, Chiarion-Sileni V, Gonzalez R,
Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M,
Rutkowski P, et al: Combined nivolumab and ipilimumab or
monotherapy in untreated melanoma. N Engl J Med. 373:23–34. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brahmer J, Reckamp KL, Baas P, Crinò L,
Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE,
Holgado E, et al: Nivolumab versus docetaxel in advanced
squamous-cell non-small-cell lung cancer. N Engl J Med.
373:123–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Motzer RJ, Rini BI, McDermott DF, Redman
BG, Kuzel TM, Harrison MR, Vaishampayan UN, Drabkin HA, George S,
Logan TF, et al: Nivolumab for metastatic renal cell carcinoma:
Results of a randomized phase II trial. J Clin Oncol. 33:1430–1437.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li X, Hu W, Zheng X, Zhang C, Du P, Zheng
Z, Yang Y, Wu J, Ji M, Jiang J, et al: Emerging immune checkpoints
for cancer therapy. Acta Oncol. 54:1706–1713. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Le Mercier I, Lines JL and Noelle RJ:
Beyond CTLA-4 and PD-1, the generation Z of negative checkpoint
regulators. Front Immunol. 6:4182015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nomi T, Sho M, Akahori T, Hamada K, Kubo
A, Kanehiro H, Nakamura S, Enomoto K, Yagita H, Azuma M, et al:
Clinical significance and therapeutic potential of the programmed
death-1 ligand/programmed death-1 pathway in human pancreatic
cancer. Clin Cancer Res. 13:2151–2157. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Okudaira K, Hokari R, Tsuzuki Y, Okada Y,
Komoto S, Watanabe C, Kurihara C, Kawaguchi A, Nagao S, Azuma M, et
al: Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a
mouse pancreatic cancer model. Int J Oncol. 35:741–749.
2009.PubMed/NCBI
|
|
17
|
Brahmer JR, Drake CG, Wollner I, Powderly
JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller
TL, et al: Phase I study of single-agent anti-programmed death-1
(MDX-1106) in refractory solid tumors: Safety, clinical activity,
pharmacodynamics, and immunologic correlates. J Clin Oncol.
28:3167–3175. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ,
Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al:
Safety and activity of anti-PD-L1 antibody in patients with
advanced cancer. N Engl J Med. 366:2455–2465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Le DT, Lutz E, Uram JN, Sugar EA, Onners
B, Solt S, Zheng L, Diaz LA Jr, Donehower RC, Jaffee EM, et al:
Evaluation of ipilimumab in combination with allogeneic pancreatic
tumor cells transfected with a GM-CSF gene in previously treated
pancreatic cancer. J Immunother. 36:382–389. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G,
Soares K, Solt S, Dorman A, Wamwea A, Yager A, et al: Immunotherapy
converts nonimmunogenic pancreatic tumors into immunogenic foci of
immune regulation. Cancer Immunol Res. 2:616–631. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang P, Su DM, Liang M and Fu J:
Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1)
surface expression in breast cancer cells and promote
PD-L1-mediated T cell apoptosis. Mol Immunol. 45:1470–1476. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ghebeh H, Lehe C, Barhoush E, Al-Romaih K,
Tulbah A, Al-Alwan M, Hendrayani SF, Manogaran P, Alaiya A,
Al-Tweigeri T, et al: Doxorubicin downregulates cell surface B7-H1
expression and upregulates its nuclear expression in breast cancer
cells: Role of B7-H1 as an anti-apoptotic molecule. Breast Cancer
Res. 12:R482010. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gong W, Song Q, Lu X, Gong W, Zhao J, Min
P and Yi X: Paclitaxel induced B7-H1 expression in cancer cells via
the MAPK pathway. J Chemother. 23:295–299. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peng J, Hamanishi J, Matsumura N, Abiko K,
Murat K, Baba T, Yamaguchi K, Horikawa N, Hosoe Y, Murphy SK, et
al: Chemotherapy induces programmed cell death-ligand 1
overexpression via the nuclear factor-κB to foster an
immunosuppressive tumor microenvironment in ovarian cancer. Cancer
Res. 75:5034–5045. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Higashimura Y, Naito Y, Takagi T,
Mizushima K, Hirai Y, Harusato A, Ohnogi H, Yamaji R, Inui H,
Nakano Y, et al: Oligosaccharides from agar inhibit murine
intestinal inflammation through the induction of heme oxygenase-1
expression. J Gastroenterol. 48:897–909. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanda Y: Investigation of the freely
available easy-to-use software ‘EZR’ for medical statistics. Bone
Marrow Transplant. 48:452–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI,
Park YM, Oh S, Shin JG, Yao S, Chen L, et al: Interferon regulatory
factor-1 is prerequisite to the constitutive expression and
IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett.
580:755–762. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mimura K, Kua LF, Shiraishi K, Kee Siang
L, Shabbir A, Komachi M, Suzuki Y, Nakano T, Yong WP, So J, et al:
Inhibition of mitogen-activated protein kinase pathway can induce
upregulation of human leukocyte antigen class I without
PD-L1-upregulation in contrast to interferon-γ treatment. Cancer
Sci. 105:1236–1244. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee SK, Seo SH, Kim BS, Kim CD, Lee JH,
Kang JS, Maeng PJ and Lim JS: IFN-gamma regulates the expression of
B7-H1 in dermal fibroblast cells. J Dermatol Sci. 40:95–103. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Isomura I, Shintani Y, Yasuda Y, Tsujimura
K and Morita A: Induction of regulatory dendritic cells by topical
application of NF-kappaB decoy oligodeoxynucleotides. Immunol Lett.
119:49–56. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qin X, Liu C, Zhou Y and Wang G: Cisplatin
induces programmed death-1-ligand 1(PD-L1) over-expression in
hepatoma H22 cells via Erk/MAPK signaling pathway. Cell Mol Biol.
Suppl 56:OL1366–OL1372. 2010.PubMed/NCBI
|
|
32
|
Maude SL, Frey N, Shaw PA, Aplenc R,
Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et
al: Chimeric antigen receptor T cells for sustained remissions in
leukemia. N Engl J Med. 371:1507–1517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bocci G, Danesi R, Di Paolo AD, Innocenti
F, Allegrini G, Falcone A, Melosi A, Battistoni M, Barsanti G,
Conte PF, et al: Comparative pharmacokinetic analysis of
5-fluorouracil and its major metabolite 5-fluoro-5,6-dihydrouracil
after conventional and reduced test dose in cancer patients. Clin
Cancer Res. 6:3032–3037. 2000.PubMed/NCBI
|
|
34
|
Kroep JR, Giaccone G, Voorn DA, Smit EF,
Beijnen JH, Rosing H, van Moorsel CJ, van Groeningen CJ, Postmus
PE, Pinedo HM, et al: Gemcitabine and paclitaxel: Pharmacokinetic
and pharmacodynamic interactions in patients with non-small-cell
lung cancer. J Clin Oncol. 17:2190–2197. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sakai H, Kokura S, Ishikawa T, Tsuchiya R,
Okajima M, Matsuyama T, Adachi S, Katada K, Kamada K, Uchiyama K,
et al: Effects of anticancer agents on cell viability,
proliferative activity and cytokine production of peripheral blood
mononuclear cells. J Clin Biochem Nutr. 52:64–71. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Okino H, Maeyama R, Manabe T, Matsuda T
and Tanaka M: Trans-tissue, sustained release of gemcitabine from
photocured gelatin gel inhibits the growth of heterotopic human
pancreatic tumor in nude mice. Clin Cancer Res. 9:5786–5793.
2003.PubMed/NCBI
|
|
37
|
Pardoll DM: The blockade of immune
checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Parsa AT, Waldron JS, Panner A, Crane CA,
Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et
al: Loss of tumor suppressor PTEN function increases B7-H1
expression and immunoresistance in glioma. Nat Med. 13:84–88. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Marzec M, Zhang Q, Goradia A, Raghunath
PN, Liu X, Paessler M, Wang HY, Wysocka M, Cheng M, Ruggeri BA, et
al: Oncogenic kinase NPM/ALK induces through STAT3 expression of
immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci
USA. 105:20852–20857. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Deer EL, González-Hernández J, Coursen JD,
Shea JE, Ngatia J, Scaife CL, Firpo MA and Mulvihill SJ: Phenotype
and genotype of pancreatic cancer cell lines. Pancreas. 39:425–435.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Awasthi N, Zhang C, Schwarz AM, Hinz S,
Wang C, Williams NS, Schwarz MA and Schwarz RE: Comparative
benefits of Nab-paclitaxel over gemcitabine or polysorbate-based
docetaxel in experimental pancreatic cancer. Carcinogenesis.
34:2361–2369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Holcomb B, Yip-Schneider MT, Matos JM,
Dixon J, Kennard J, Mahomed J, Shanmugam R, Sebolt-Leopold J and
Schmidt CM: Pancreatic cancer cell genetics and signaling response
to treatment correlate with efficacy of gemcitabine-based molecular
targeting strategies. J Gastrointest Surg. 12:288–296. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Adachi S, Kokura S, Okayama T, Ishikawa T,
Takagi T, Handa O, Naito Y and Yoshikawa T: Effect of hyperthermia
combined with gemcitabine on apoptotic cell death in cultured human
pancreatic cancer cell lines. Int J Hyperthermia. 25:210–219. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cao LP, Song JL, Yi XP and Li YX: Double
inhibition of NF-κB and XIAP via RNAi enhances the sensitivity of
pancreatic cancer cells to gemcitabine. Oncol Rep. 29:1659–1665.
2013.PubMed/NCBI
|
|
45
|
Thoennissen NH, Iwanski GB, Doan NB,
Okamoto R, Lin P, Abbassi S, Song JH, Yin D, Toh M, Xie WD, et al:
Cucurbitacin B induces apoptosis by inhibition of the JAK/STAT
pathway and potentiates antiproliferative effects of gemcitabine on
pancreatic cancer cells. Cancer Res. 69:5876–5884. 2009. View Article : Google Scholar : PubMed/NCBI
|