Introduction
Acute lymphoblastic leukemia (ALL) is a malignant
disorder of lymphoid progenitor cells. The steady progress in the
development of effective treatments has led to a cure rate of more
than 80% in children with ALL, most of whom will lead healthy
productive lives as long-term cancer survivors (1,2). Yet,
studies are underway to ascertain the precise events that take
place in the genesis of ALL, to enhance the clinical application of
known risk factors and anti-leukemic agents, and to identify
treatment regimens that may boost the generally low cure rates in
adults and subgroups of children with high-risk leukemia (3–5).
Approaches aimed at blocking associated miRNAs may serve as
effective therapeutic strategies for treating ALL patients and may
be valuable biomarkers for diagnosis and treatment.
Dysregulated Wnt signaling has been reported as a
hallmark of particular types of solid tumors (6–9).
Several studies have also indicated that Wnt signal transduction
plays a key role in certain stages of lymphocyte development and
the self-renewal of hematopoietic stem cells (10–16),
suggesting its dysregulation as a mechanism underlying lymphoid
leukemogenesis. Recent studies concerning acute or chronic lymphoid
leukemias have provided further evidence for the role of Wnt
signaling in malignant hematopoiesis (10–12,17,18).
The deficiency of Wnt antagonists can contribute to
activation of the Wnt pathway resulting in carcinogenesis via
deregulation of cell proliferation and differentiation. Numerous
studies have shown that impaired regulation of Wnt antagonists such
as Wnt inhibitory factor-1 (WIF1), sFRP, HDPR1 and DKK3 by promoter
hypermethylation is present in several human malignancies including
ALL (17–22). However, it is noteworthy that the
frequency of methylation of WIF1 is less than 30% in ALL (20), which implies that there may be
additional regulatory mechanisms responsible for downregulating
WIF1 expression.
miRNAs are short (19–25 nucleotides) RNA molecules
that can modulate the expression of a wide range of target genes by
pairing homologous sequences within the 3′-UTR of mRNAs, thus
impairing their translation or promoting RNA degradation (23,24) in
multiple diseases including cancers. miR-181a has been proposed to
play multi-roles in neoplasia and progression. miR-181a-5p is an
oncogenic miRNA found to be deregulated in multiple types of tumors
[e.g., breast cancer (25–27), osteosarcoma (28), colorectal carcinoma (29), gastric cancer (30), salivary adenoid cystic carcinoma
(31) and lung cancer (32)]. In contrast, miR-181a was
downregulated in many other tumors and thus served as a
tumor-suppressor gene (33,34). In previous studies of hematologic
malignancies, miR-181a was found to be upregulated in acute myeloid
leukemia (35) and myelodysplastic
syndromes (36), but downregulated
in multiple myeloma (37) and
chronic lymphocyte leukemia (38).
In this study, we demonstrated that upregulation of
miR-181a-5p directly targets WIF1 in ALL cells, suggesting that
miR-181a-5p-mediated Wnt-signaling activation may be implicated in
the pathogenesis of ALL.
Materials and methods
Cell lines and clinical samples
Two ALL-derived cell lines (Jurkat and MOLT-4) and
other hematopoietic tumor cell lineages (HL60, NB4 and KG-1) were
obtained from the Cancer Research Institute, Southern Medical
University, Guangzhou, China. Cells were cultured at 37°C under 5%
CO2 in humidified air in RPMI-1640 medium (HyClone,
Logan, UT, USA) supplemented with 10% fetal bovine serum (Gibco,
Grand Island, NY, USA). Thirty-seven primary ALL patients and 28
ALL-complete remission bone marrow samples were collected from The
Third Affiliated Hospital and Zhujiang Hospital, Southern Medical
University. Twenty-three samples of normal peripheral blood
mononuclear cells (PBMCs) were obtained from Guangzhou Blood
Center.
The study was approved by the Human Ethics Committee
at The Third Affiliated Hospital of Southern Medical University.
Written informed consent was obtained from all participants.
Extraction of total RNA and
quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from tissues and cell lines
with TRIzol (Invitrogen, Carlsbad, CA, USA) according to the user
manual. For mRNA expression analysis, 1 µg of total RNA was used
for RT using PrimerScript™ RT reagent kit following the
manufacturer's instructions (lot. no. BK3001), and real-time PCR
was performed using SYBR® Premix Ex Taq™ Real-Time PCR
kit (code no. DRR041A) (both from Takara Bio) on an Mx3005P
Stratagene. All data were normalized to GAPDH expression and
further normalized to the negative control unless otherwise
indicated. Primer sequences for WIF-1 (forward,
5′-TCTCCAAACACCTCAAAATGCT-3′ and reverse,
5′-GACACTCGCAGATGCGTCT-3′) and GAPDH (forward,
5′-CCATGAGAAGTATGACAACAGCC-3′ and reverse,
5′-GGGTGCTAAGCAGTTGGTG-3′) were directly acquired from PrimerBank
(http://pga.mgh.harvard.edu/primerbank/). For miRNA
expression analysis, mature miRNAs were reverse-transcribed, and
real-time PCR was performed using All-in-One™ miRNA qRT-PCR
detection kit following the manufacturer's instructions (cat. no.
AOMD-Q020; GeneCopoeia, Inc., Rockville, MD, USA). All data were
normalized to U6 expression. The fold changes were
calculated by relative quantification (2−∆∆Ct). qRT-PCR
was conducted for each sample in triplicate.
MTT assay
MOLT-4 (5×103) and Jurkat cells
(3×103) were plated onto 96-well plates (NEST
Biotechnology, Wuxi, China) respectively in 100 µl growth medium
and allowed to adhere overnight. The cells were then transfected
with 50 nM of miRNA mimics (GenePharma, Shanghai, China) or siRNA
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and inhibitor
(GenePharma), respectively. At different time-points (24, 48 and 72
h), the culture medium was removed and replaced with culture medium
containing 10 µl of sterile
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT) dye (5 mg/ml). After incubation at 37°C for 4 h, the MTT
solution was removed, and 150 µl dimethyl sulfoxide (DMSO) was
added to dissolve the formazan crystals. Spectrometric absorbance
at 490 nm was measured using a BioTek ELx800 microplate photometer
(BioTek ELx800, SN211805; BioTek, Winooski, VT, USA).
Anchorage-independent growth
assay
Cells were blowed gently, and 1×105 cells
were resuspended in 2 ml complete medium plus 0.3% agar (Sigma, St.
Louis, MO, USA). The agar-cell mixture was plated on top of a
bottom layer consisting of 1% agar in complete medium. After 15
days, colony size was measured using an ocular micrometer and
colonies larger than 0.1 mm in diameter were counted. The
experiment was performed three times for each cell line.
5-Ethynyl-2′-deoxyuridine assay
Cells were transfected with miRNA mimics in 96-well
plates. Forty-eight hours after transfection,
5-ethynyl-2′-deoxyuridine (EdU) (100 µM) (Cell Light EdU DNA
imaging kit; Guangzhou RiboBio Co., Ltd., Guangzhou, China) was
added, and the cells were cultured for an additional 2 h. The cells
were then stained according to the production manual (Guangzhou
RiboBio Co., Ltd.). Images were captured and analyzed using
fluorescence microscopy (Nikon Eclipse 80i; Nikon, Tokyo, Japan).
EdU-positive cells were determined with the formula: (EdU-treated
cells/DAPI stained cells) × 100%.
Flow cytometry
The cells were fixed in 70% ice-cold ethanol for 48
h at 4°C and stained by incubation with PBS containing 10 µg/ml
propidium iodide and 0.5 mg/ml RNaseA for 15 min at 37°C. The cells
were analyzed for the DNA content of labeled cells by FACSCaliber
cytometry (BD Biosciences, Franklin Lakes, NJ, USA). Each
experiment was conducted in triplicate.
Dual-luciferase assay
The luciferase reporter constructs pEZX-MT01-WIF1
3′-UTR WT and miRNA target clone control vector for pEZX-MT01 were
purchased from GeneCopoeia, Inc. (HmiT001390-MT01 and CmiT000001-
MT01). Mutant reporter plasmids were obtained from this plasmid
using a KOD-Plus Mutagenesis kit (SMK-101; Toyobo Co., Ltd., Life
Science Department, Osaka, Japan). Luciferase assays were conducted
using 293T cells plated in a 24-well plate (NEST Biotechnology).
Transfections were performed using Lipofectamine™ 2000 (11668–019;
Invitrogen) in Opti-MEM serum-free media (cat. no. 10742; Gibco).
Luciferase and Renilla signals were measured 48 h after
transfection using the Dual-Luciferase Reporter Assay kit (Promega
Corp., Madison, WI, USA) according to the manufacturer's
instructions. Three independent experiments were performed, and the
data are presented as the mean ± SD.
Western blot assay
Cell lysate was prepared using RIPA buffer with
protease inhibitors and quantified using the BCA protein assay
(BioTek China, Beijing, China). The Nuclear Protein Extraction kit
(BSP009; Sangon Biotech Co., Ltd., Shanghai, China) was used for
extracting nuclear proteina of the ALL cells. Protein (20 µg) was
loaded onto a 10% SDS-PAGE gel that was then transferred onto a
PVDF membrane and incubated with anti-WIF1 (1:1,000, ab2064; Cell
Signaling Technology, Inc., Danvers, MA, USA), anti-C-myc
(1:500, SC-40), anti-E2F1 (1:1,000, SC-22820),
anti-CDK4 (1:1,000, SC-260), anti-CCND1 (1:500,
SC-753) and anti-P21 (1:1,000, SC-397) (all from Santa Cruz
Biotechnology, Inc.) at 4°C overnight in blocker (3% non-fat dry
milk/BSA in TTBS) followed by incubation with HRP-conjugated
secondary anti-mouse antibody (1:2,000, ZB2305; ZSGB-BIO, Beijing,
China). Protein was normalized with GAPDH (1:5,000, no.
P30008; Abmart, Shanghai, China) and histone-H3 (1:2,000,
17168-1-AP; ProteinTech Group, Inc., Chicago, IL, USA).
Statistical analysis
All statistical analyses were performed using the
SPSS 13.0 Statistical Software Package (SPSS, Inc., Chicago, IL,
USA). Two-tailed Student's t-test was used to determine the
differences between groups for in vitro and in vivo
analyses. The differences were considered to be statistically
significant at a P-value <0.05. All data are presented as mean ±
SD or SEM unless otherwise noted.
Results
miR-181a-5p is highly expressed in
various leukemia cell lines and primary ALL
RT-qPCR analyses initially showed that the
expression of miR-181a-5p was markedly upregulated in 5 cell lines
(HL60, MOLT-4, JURKAT, NB4 and KG-1) as compared with that in the
normal PBMCs from healthy voluntary individuals (Fig. 1A). To validate the clinical
relevance of miR-181a-5p to ALL patients, we further examined the
miR-181a-5p expression level in 37 primary ALL samples and 28
ALL-complete remission samples compared with 23 PBMC samples. As
shown in Fig. 1B, miR-181a-5p was
highly expressed in the primary ALL patient samples. Notably, the
expression level of miR-181a-5p was significantly reduced in the
ALL-complete remission patient samples compared with primary ALL
patients. These data suggest that miR-181a-5p may play an important
role in the pathogenesis of ALL, and is associated with prognosis
and may be valuable in the evaluation of therapeutic effects.
miR-181a-5p promotes ALL cell growth
and proliferation in vitro
To explore the role of miR-181a-5p in the
development and progression of ALL, we next examined its role in
cellular proliferation. MOLT-4 cells with lower miR-181a-5p
expression were used in the gain-of-function studies, whereas
Jurkat cells with higher miR-181a-5p expression (Fig. 1A) were applied in the
loss-of-function analyses. MTT and soft agar colony formation
assays were conducted in the MOLT-4 cells transfected with the
miR-181a-5p mimic and in Jurkat cells transfected with the
miR-181a-5p inhibitor. The miR-181a-5p mimic increased cell growth
and anchorage-independent growth ability of the MOLT-4 cells
(Fig. 2A and B), while the
miR-181a-5p inhibitor markedly reduced cell growth and
anchorage-independent growth ability of the Jurkat cells (Fig. 3A and B).
Using flow cytometric analysis, we further found
that MOLT-4 cells transfected with the miR-181a-5p mimic exhibited
a significantly reduced cell proportion in the G1 phase by
9.22±0.34% (P<0.001) and increased proliferation (S/G2/M cell
proportion) by 9.18±0.30% (P<0.001) (Fig. 2C), whereas the miR-181a-5p inhibitor
increased the percentage of cells in the G1 phase by 9.02±0.83%
(P<0.001) and reduced the proliferation of Jurkat cells (S/G2/M
cell proportion) by 9.08±0.47% (P<0.001) (Fig. 3C). Consistently, EdU incorporation
assay showed that the percentage of cells in S phase was
significantly increased in the miR-181a-5p-overexpressing MOLT-4
cells compared with the miR-control MOLT-4 cells (Fig. 2D), but the miR-181a-5p inhibitor
reduced the percentage of Jurkat cells in the S phase (Fig. 3D). These results suggest that
miR-181a-5p modulates ALL cell proliferation through regulation of
G1/S transition.
WIF1 is a major target gene of
miR-181a-5p in ALL cells
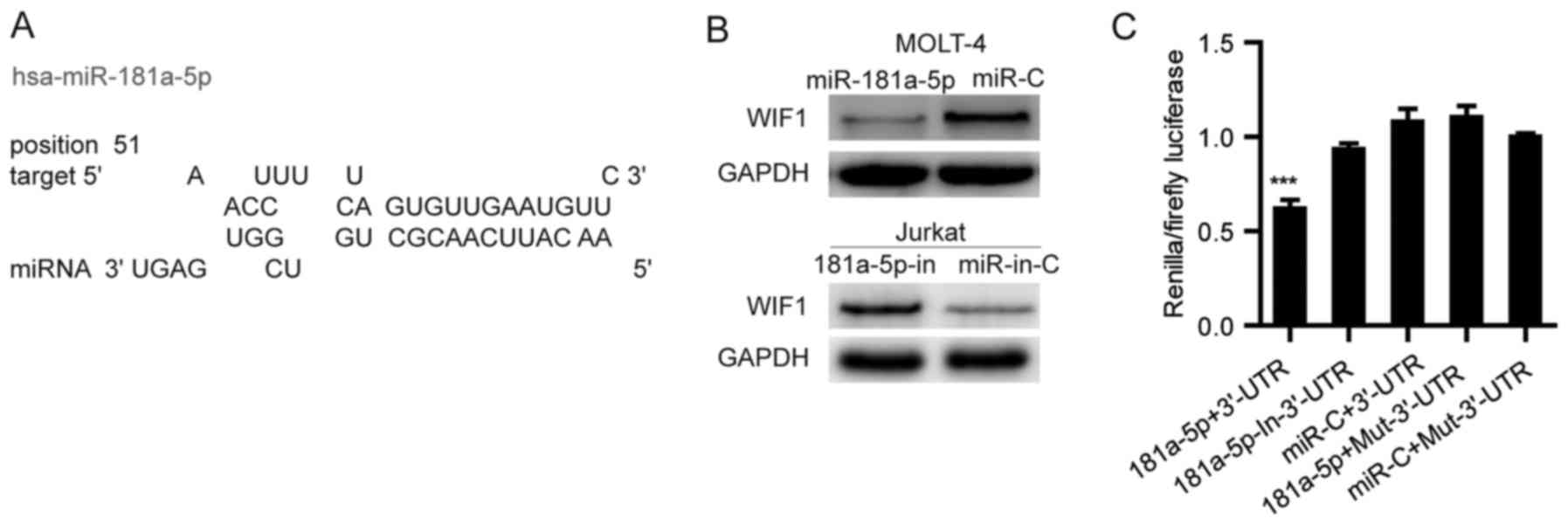
WIF1 is a Wnt antagonist that inhibits Wnt signaling
by direct binding to Wnt molecules. Using miRwalk, a publicly
available algorithm, we found that WIF1 is theoretically the target
gene of miR-181a-5p (Fig. 4A and
Table I). To validate whether WIF1
is a direct target of miR-181a-5p, a wild-type or mutant 3′-UTR
fragment of WIF1 was cloned downstream of the firefly luciferase
gene. Dual-luciferase reporter assays revealed that miR-181a-5p
significantly attenuated the activity of firefly luciferase with
the wild-type 3′-UTR of WIF1, whereas this effect was abolished
when the predicted 3′-UTR-binding site was mutated (Fig. 4B). Subsequent western blot analysis
confirmed that miR-181a-5p attenuated the expression of cellular
WIF1 in MOLT-4 cells and silencing of miR-181a-5p increased the
level of cellular endogenous WIF1 protein in the Jurkat cells
(Fig. 4C). These data suggest that
miR-181a-5p may inhibit the expression of WIF1 by directly binding
to its 3′-UTR.
 | Table I.Predicted microRNA according to mRNA
selected regions (minimum seed length, 7; P<0.001). |
Table I.
Predicted microRNA according to mRNA
selected regions (minimum seed length, 7; P<0.001).
| microRNA | Gene | RefseqID | Seed length | Start | Seed sequence | End | P-value |
|---|
| hsa-miR-181a | CPOX | NM_000097 | 11 | 2596 | AACAUUCAACG | 2586 | 0.0003 |
| hsa-miR-181a | CHST9 | NM_031422 | 10 | 3110 | AACAUUCAAC | 3101 | 0.0006 |
| hsa-miR-181a | HMGB2 | NM_002129 | 10 | 1055 | AACAUUCAAC | 1046 | 0.0007 |
| hsa-miR-181a | IL2 | NM_000586 | 10 | 739 | AACAUUCAAC | 730 | 0.0003 |
| hsa-miR-181a | SYNE1 | NM_182961 | 10 | 27647 | AACAUUCAAC | 27638 | 0.0007 |
|
hsa-miR-181a | WIF1 |
NM_007191 | 10 | 1357 |
AACAUUCAAC | 1348 | 0.0007 |
| hsa-miR-181a | C12orf56 | NM_001099676 | 9 | 1529 | AACAUUCAA | 1521 | 0.0009 |
| hsa-miR-181a | DNAJC3 | NM_006260 | 9 | 5051 | ACAUUCAAC | 5043 | 0.0000 |
| hsa-miR-181a | ELAVL4 | NM_021952 | 9 | 1664 | AACAUUCAA | 1656 | 0.0009 |
| hsa-miR-181a | HIGD2A | NM_138820 | 9 | 548 | AACAUUCAA | 540 | 0.0010 |
| hsa-miR-181a | PIH1D2 | NM_001082619 | 9 | 1095 | AACAUUCAA | 1087 | 0.0007 |
| hsa-miR-181a | SFT2D1 | NM_145169 | 9 | 546 | AACAUUCAA | 538 | 0.0008 |
| hsa-miR-181a | CENPI | NM_006733 | 8 | 2550 | AACAUUCA | 2543 | 0.0005 |
| hsa-miR-181a | KRTCAP2 | NM_173852 | 8 | 526 | AACAUUCA | 519 | 0.0009 |
| hsa-miR-181a | NCR2 | NM_004828 | 8 | 940 | AACAUUCA | 933 | 0.0004 |
| hsa-miR-181a | WDR79 | NM_018081 | 8 | 1836 | ACAUUCAA | 1829 | 0.0002 |
WIF1 suppression is required for
miR-181a-5p-induced ALL cell proliferation
To further confirm the role of WIF1 suppression in
miR-181a-5p-induced ALL cell proliferation, we knocked down WIF1
expression in ALL cells (MOLT-4) using specific siRNA (Fig. 5E) and then observed the alteration
of cell proliferation. Similar to miR-181a-5p, WIF1 siRNA enabled
an obviously reduced WIF1 expression followed by increased ALL cell
growth and proliferation (Fig.
5A-E). In addition, following treatment of Jurkat cells with
the miR-181a-5p inhibitor, significantly increased WIF1 expression
and restricted cell proliferation were observed. Furthermore, we
transfected WIF1-siRNA into the miR-181a-5p inhibitor-treated cells
and observed that G1/S phase cell cycle arrest due to miR-181a-5p
inhibition was abrogated (Fig. 5F and
G). Taken together, these data suggest that WIF1 suppression is
required for miR-181a-5p-induced ALL cell proliferation.
miR-181a-5p activates Wnt/β-catenin
signaling through suppression of WIF1
The cellular fractionation and western blotting
showed that miR-181a-5p overexpression promoted nuclear
accumulation of β-catenin, indicating that miR-181a-5p may activate
the Wnt/β-catenin pathway through promoting nuclear β-catenin
accumulation (Fig. 6A).
Accordingly, we investigated whether miR-181a-5p influences
downsteam signaling of the Wnt/β-catenin pathway. As expected, the
expression levels of c-myc, E2F1, cyclin D1, and CDK4 were
increased while p21 expression was decreased following
overexpression of miR-181a-5p. Opposite results were found in the
miR-181a-5p-inhibited ALL cells (Fig.
6B).
Discussion
The expression and functions of specific miRNAs
generally differ distinctly depending on the cancer cell type. In
the present study, we observed that miR-181a-5p expression was
highly expressed in several leukemia cell lines compared with that
in normal PBMCs. Notably, we found that miR-181a-5p was
downregulated in ALL-complete remission samples compared with that
in primary ALL samples, indicating that miR-181a-5p may be a
potential biomarker for the evaluation of clinical efficacy and
prognosis of ALL. This is consistent with a previous study that
increased expression of miR-181a is associated with a poor outcome
of ALL (39).
miR-181a can serve as an oncomiR or tumor
suppressor, also implicating its complexity and muti-functions in
the regulation of its target genes or signaling pathways in cancer.
In the present study, we found that the ectopic expression of
miR-181a enhanced ALL cell proliferation and silencing of miR-181a
expression displayed the opposite effect, suggesting that abnormal
miR-181a expression may contribute to the pathogenesis of ALL.
Furthermore, we used a bioinformatics approach to predict the
target genes of hsa-miR-181a-5p. Among the potential candidates,
WIF1 attracted our attention since it is a classic tumor-suppressor
gene involved in the cell proliferation in various cancers
(40–42). Importantly, Wnt/β-catenin signaling
is frequently activated in ALL (43,44),
but its precise mechanism of action is not well understood. WIF1
has proved to act as a secreted Wnt inhibitor that binds directly
to Wnt, and thus constraining the binding of Wnt ligands to the
Frizzled receptor (45). Thus, the
downregulation of WIF1 contributes to activation of the Wnt pathway
resulting in carcinogenesis through dysregulation of cell
proliferation and differentiation. Luciferase reporter assays and
western blotting further confirmed that miR-181a-5p directly
suppressed WIF1 expression via directly targeting the 3′-UTR of
WIF1. Although it has been reported that WIF1 is a target gene of
miR-181a in colorectal cancer (46), our study suggests that WIF1 is a
major target of miR-181a-5p and inactivation induced by miR-181a-5p
may play an important role in promoting the carcinogenesis of
ALL.
β-catenin is a major cellular effector of Wnt
signaling. It is normally targeted by a complex of axin, APC,
glycogen synthase kinase-3β and casein kinase 1α for
proteasome-mediated degradation following phosphorylation and
ubiquitination (47,48). β-catenin accumulation and nuclear
translocation to regulate genes are important for cell
proliferation. Notably, we observed that high expression of
miR-181a-5p led to increased nuclear accumulation of β-catenin,
indicating that miR-181a-5p can activate Wnt/β-catenin signaling in
ALL. Subsequently, we detected the downstream targets of activated
Wnt pathway signaling, confirming the activation of Wnt/β-catenin
signaling in the presence of WIF1 inactivation via miR-181a-5p in
ALL. Therefore, in vivo antagomir-based strategies may be
researched for further understanding the basic and translational
significance of miR-181a-5p-Wnt/β-catenin signaling in ALL.
In conclusion, we demonstrated that WIF1 is a major
target of miR-181a-5p in ALL and downregulation induced by
miR-181a-5p activated Wnt/β-catenin signaling, promoting the
proliferation of ALL. These findings uncover a crucial molecular
mechanism that maintains the constitutive activation of the
Wnt/β-catenin pathway and may prove to be clinically useful for
developing a new curative and effective biomarker and therapeutic
target for ALL.
Acknowledgements
This study was jointly funded by grants from the
National Natural Science Foundation of China (no. 81502335) and
Natural Science Foundation of Guangdong Province (no.
S2014A030310028).
References
|
1
|
Pui CH and Evans WE: Treatment of acute
lymphoblastic leukemia. N Engl J Med. 354:166–178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Flotho C, Coustan-Smith E, Pei D, Cheng C,
Song G, Pui CH, Downing JR and Campana D: A set of genes that
regulate cell proliferation predicts treatment outcome in childhood
acute lymphoblastic leukemia. Blood. 110:1271–1277. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jabeen K, Ashraf MS, Iftikhar S and
Belgaumi AF: The impact of socioeconomic factors on the outcome of
childhood acute lymphoblastic leukemia (ALL) treatment in a
low/middle income country (LMIC). J Pediatr Hematol Oncol.
38:587–596. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dinmohamed AG, Szabó A, van der Mark M,
Visser O, Sonneveld P, Cornelissen JJ, Jongen-Lavrencic M and
Rijneveld AW: Improved survival in adult patients with acute
lymphoblastic leukemia in the Netherlands: A population-based study
on treatment, trial participation and survival. Leukemia.
30:310–317. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cruz-Rodriguez N, Combita AL, Enciso LJ,
Quijano SM, Pinzon PL, Lozano OC, Castillo JS, Li L, Bareño J,
Cardozo C, et al: High expression of ID family and IGJ genes
signature as predictor of low induction treatment response and
worst survival in adult Hispanic patients with B-acute
lymphoblastic leukemia. J Exp Clin Cancer Res. 35:642016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
van Es JH, Barker N and Clevers H: You Wnt
some, you lose some: Oncogenes in the Wnt signaling pathway. Curr
Opin Genet Dev. 13:28–33. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee HC, Kim M and Wands JR: Wnt/Frizzled
signaling in hepatocellular carcinoma. Front Biosci. 11:1901–1915.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim M, Lee HC, Tsedensodnom O, Hartley R,
Lim YS, Yu E, Merle P and Wands JR: Functional interaction between
Wnt3 and Frizzled-7 leads to activation of the Wnt/beta-catenin
signaling pathway in hepatocellular carcinoma cells. J Hepatol.
48:780–791. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wei W, Chua MS, Grepper S and So SK:
Soluble Frizzled-7 receptor inhibits Wnt signaling and sensitizes
hepatocellular carcinoma cells towards doxorubicin. Mol Cancer.
10:162011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reya T, Duncan AW, Ailles L, Domen J,
Scherer DC, Willert K, Hintz L, Nusse R and Weissman IL: A role for
Wnt signalling in self-renewal of haematopoietic stem cells.
Nature. 423:409–414. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weerkamp F, vanDongen JJ and Staal FJ:
Notch and Wnt signaling in T-lymphocyte development and acute
lymphoblastic leukemia. Leukemia. 20:1197–1205. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khan NI and Bendall LJ: Role of WNT
signaling in normal and malignant hematopoiesis. Histol
Histopathol. 21:761–774. 2006.PubMed/NCBI
|
|
13
|
Staal FJ and Clevers HC: WNT signalling
and haematopoiesis: A WNT-WNT situation. Nat Rev Immunol. 5:21–30.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Døsen G, Tenstad E, Nygren MK, Stubberud
H, Funderud S and Rian E: Wnt expression and canonical Wnt
signaling in human bone marrow B lymphopoiesis. BMC Immunol.
7:132006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Staal FJ and Sen JM: The canonical Wnt
signaling pathway plays an important role in lymphopoiesis and
hematopoiesis. Eur J Immunol. 38:1788–1794. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Timm A and Grosschedl R: Wnt signaling in
lymphopoiesis. Curr Top Microbiol Immunol. 290:225–252.
2005.PubMed/NCBI
|
|
17
|
Batra S, Shi Y, Kuchenbecker KM, He B,
Reguart N, Mikami I, You L, Xu Z, Lin YC, Clément G, et al: Wnt
inhibitory factor-1, a Wnt antagonist, is silenced by promoter
hypermethylation in malignant pleural mesothelioma. Biochem Biophys
Res Commun. 342:1228–1232. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu TH, Raval A, Chen SS, Matkovic JJ,
Byrd JC and Plass C: CpG island methylation and expression of the
secreted frizzled-related protein gene family in chronic
lymphocytic leukemia. Cancer Res. 66:653–658. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yau TO, Chan CY, Chan KL, Lee MF, Wong CM,
Fan ST and Ng IO: HDPR1, a novel inhibitor of the WNT/beta-catenin
signaling, is frequently downregulated in hepatocellular carcinoma:
Involvement of methylation-mediated gene silencing. Oncogene.
24:1607–1614. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roman-Gomez J, Jimenez-Velasco A, Agirre
X, Castillejo JA, Navarro G, Barrios M, Andreu EJ, Prosper F,
Heiniger A and Torres A: Transcriptional silencing of the
Dickkopfs-3 (Dkk-3) gene by CpG hypermethylation in acute
lymphoblastic leukaemia. Br J Cancer. 91:707–713. 2004.PubMed/NCBI
|
|
21
|
Suzuki H, Watkins DN, Jair KW, Schuebel
KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Van
Engeland M, et al: Epigenetic inactivation of SFRP genes allows
constitutive WNT signaling in colorectal cancer. Nat Genet.
36:417–422. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mazieres J, He B, You L, Xu Z, Lee AY,
Mikami I, Reguart N, Rosell R, McCormick F and Jablons DM: Wnt
inhibitory factor-1 is silenced by promoter hypermethylation in
human lung cancer. Cancer Res. 64:4717–4720. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mori F, Strano S and Blandino G:
MicroRNA-181a/b: Novel biomarkers to stratify breast cancer
patients for PARPi treatment. Cell Cycle. 12:1823–1824. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Taylor MA, Sossey-Alaoui K, Thompson CL,
Danielpour D and Schiemann WP: TGF-β upregulates miR-181a
expression to promote breast cancer metastasis. J Clin Invest.
123:150–163. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bisso A, Faleschini M, Zampa F, Capaci V,
De Santa J, Santarpia L, Piazza S, Cappelletti V, Daidone M, Agami
R, et al: Oncogenic miR-181a/b affect the DNA damage response in
aggressive breast cancer. Cell Cycle. 12:1679–1687. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jianwei Z, Fan L, Xiancheng L, Enzhong B,
Shuai L and Can L: MicroRNA 181a improves proliferation and
invasion, suppresses apoptosis of osteosarcoma cell. Tumour Biol.
34:3331–3337. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nishimura J, Handa R, Yamamoto H, Tanaka
F, Shibata K, Mimori K, Takemasa I, Mizushima T, Ikeda M, Sekimoto
M, et al: microRNA-181a is associated with poor prognosis of
colorectal cancer. Oncol Rep. 28:2221–2226. 2012.PubMed/NCBI
|
|
30
|
Zhang X, Nie Y, Du Y, Cao J, Shen B and Li
Y: MicroRNA-181a promotes gastric cancer by negatively regulating
tumor suppressor KLF6. Tumour Biol. 33:1589–1597. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
He Q, Zhou X, Li S, Jin Y, Chen Z, Chen D,
Cai Y, Liu Z, Zhao T and Wang A: MicroRNA-181a suppresses salivary
adenoid cystic carcinoma metastasis by targeting MAPK-Snai2
pathway. Biochim Biophys Acta. 1830:5258–5266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou JY, Ma WL, Fei J, Ding DP, Shi R,
Jiang L and Zheng WL: Effects of microRNA miR-181a on gene
expression profiles of K562 cells. Nan Fang Yi Ke Da Xue Xue Bao.
26:606–609. 2006.(In Chinese). PubMed/NCBI
|
|
33
|
Li Y, Kuscu C, Banach A, Zhang Q,
Pulkoski-Gross A, Kim D, Liu J, Roth E, Li E, Shroyer KR, et al:
miR-181a-5p inhibits cancer cell migration and angiogenesis via
downregulation of matrix metalloproteinase-14. Cancer Res.
75:2674–2685. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shin KH, Bae SD, Hong HS, Kim RH, Kang MK
and Park NH: miR-181a shows tumor suppressive effect against oral
squamous cell carcinoma cells by downregulating K-ras. Biochem
Biophys Res Commun. 404:896–902. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Debernardi S, Skoulakis S, Molloy G,
Chaplin T, Dixon-McIver A and Young BD: MicroRNA miR-181a
correlates with morphological sub-class of acute myeloid leukaemia
and the expression of its target genes in global genome-wide
analysis. Leukemia. 21:912–916. 2007.PubMed/NCBI
|
|
36
|
Pons A, Nomdedeu B, Navarro A, Gaya A, Gel
B, Diaz T, Valera S, Rozman M, Belkaid M, Montserrat E, et al:
Hematopoiesis-related microRNA expression in myelodysplastic
syndromes. Leuk Lymphoma. 50:1854–1859. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pichiorri F, Suh SS, Ladetto M, Kuehl M,
Palumbo T, Drandi D, Taccioli C, Zanesi N, Alder H, Hagan JP, et
al: MicroRNAs regulate critical genes associated with multiple
myeloma pathogenesis. Proc Natl Acad Sci USA. 105:12885–12890.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu DX, Zhu W, Fang C, Fan L, Zou ZJ, Wang
YH, Liu P, Hong M, Miao KR, Liu P, et al: miR-181a/b significantly
enhances drug sensitivity in chronic lymphocytic leukemia cells via
targeting multiple anti-apoptosis genes. Carcinogenesis.
33:1294–1301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Y, Li Z, He C, Wang D, Yuan X, Chen J
and Jin J: MicroRNAs expression signatures are associated with
lineage and survival in acute leukemias. Blood Cells Mol Dis.
44:191–197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Roperch JP, Incitti R, Forbin S, Bard F,
Mansour H, Mesli F, Baumgaertner I, Brunetti F and Sobhani I:
Aberrant methylation of NPY, PENK, and WIF1 as a promising marker
for blood-based diagnosis of colorectal cancer. BMC Cancer.
13:5662013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Alvarez C, Tapia T, Cornejo V, Fernandez
W, Muñoz A, Camus M, Alvarez M, Devoto L and Carvallo P: Silencing
of tumor suppressor genes RASSF1A, SLIT2, and WIF1 by promoter
hypermethylation in hereditary breast cancer. Mol Carcinog.
52:475–487. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tang Y, Simoneau AR, Liao WX, Yi G, Hope
C, Liu F, Li S, Xie J, Holcombe RF, Jurnak FA, et al: WIF1, a Wnt
pathway inhibitor, regulates SKP2 and c-myc expression leading to
G1 arrest and growth inhibition of human invasive urinary bladder
cancer cells. Mol Cancer Ther. 8:458–468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nygren MK, Døsen-Dahl G, Stubberud H,
Wälchli S, Munthe E and Rian E: beta-catenin is involved in
N-cadherin-dependent adhesion, but not in canonical Wnt signaling
in E2A-PBX1-positive B acute lymphoblastic leukemia cells. Exp
Hematol. 37:225–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shao N, Zou J, Li J, Chen F, Dai J, Qu X,
Sun X, Ma D and Ji C: Hyper-activation of WNT/β-catenin signaling
pathway mediates anti-tumor effects of histone deacetylase
inhibitors in acute T lymphoblastic leukemia. Leuk Lymphoma.
53:1769–1778. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hsieh JC, Kodjabachian L, Rebbert ML,
Rattner A, Smallwood PM, Samos CH, Nusse R, Dawid IB and Nathans J:
A new secreted protein that binds to Wnt proteins and inhibits
their activities. Nature. 398:431–436. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ji D, Chen Z, Li M, Zhan T, Yao Y, Zhang
Z, Xi J, Yan L and Gu J: MicroRNA-181a promotes tumor growth and
liver metastasis in colorectal cancer by targeting the tumor
suppressor WIF-1. Mol Cancer. 13:862014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Willert K and Nusse R: Beta-catenin: A key
mediator of Wnt signaling. Curr Opin Genet Dev. 8:95–102. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|