Introduction
Worldwide, hepatocellular carcinoma (HCC) is one of
the most invasive, therapy-resistant and metastatic tumors
(1). Although various therapies are
used to improve outcomes of HCC patients, such as hepatic resection
or liver transplantation, the 5-year survival rate after diagnosis
is only approximately 30% (2). The
majority of HCC patients are not candidates for curative surgery
largely due to distant metastasis and high recurrence ratio at the
time of the diagnosis (3). Although
a number of reports have demonstrated that multiple signaling
pathways are involved in tumor metastasis, the molecular mechanisms
governing the metastatic cascades of HCC are complex, and our
current knowledge regarding these mechanisms remains limited
(4). Recently, microRNAs (miRNAs)
have been implicated as key regulators of these pathways (5).
miRNAs, an abundant class of endogenous, small,
non-coding RNAs of 22 nucleotides in length, are
post-transcriptional regulators that bind to specific cognate
sequences in the 3-untranslated region (3-UTR) of target
transcripts. These RNAs commonly result in translational repression
and gene silencing (6–8). Evidence is being accumulated which
indicates that miRNAs have been reported to be involved with
diverse biological processes, such as development, inflammation and
tumorigenesis (9). It should be
noted that numerous miRNAs play crucial roles in the regulation of
HCC tumorigenesis, such as miR-221, miR-122, miR-451, miR-452 and
miR-1180 (7,10–13).
In the present study, we investigated the possible
involvement of miRNAs in the proliferation and migration of HCC. We
found that at high expression levels, microRNA-429 (miR-429) played
a critical role in regulating NF-κB pathway activity in HCC to
inhibit tumor progression. miR-429 inhibited the NF-κB pathway by
targeting TRAF6. The targeting of TRAF6 not only reduces the
ability of HCC growth but also inhibits tumor migration and
metastasis by downregulating TCF4, pTAK1 and c-Myc in vitro
and in vivo. These findings provide new insights into the
molecular mechanisms that regulate the expansion and functions of
tumor-associated HCC and into the exploration of potential targets
for therapeutic intervention.
Materials and methods
Cell culture and reagents
HL7702, SK-HEP1, SMMC7721, HCCLM3 and HepG2 were
purchased from the Institute of Biochemistry and Cell Biology,
Chinese Academy of Sciences (Shanghai, China) and cultured, as
recommended, as monolayers in RPMI-1640 supplemented with 10% fetal
bovine serum (FBS; HyClone Laboratories, Inc., Logan, UT, USA) and
1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA) in a
humidified incubator at 37°C in a 5% CO2 atmosphere.
Tissue samples
A total of 25 pairs of human primary HCC tissues,
and their corresponding non-tumorous tissues were collected from
The Affiliated Hospital of Qingdao University, China. These HCC
cases consisted of 20 males and 5 females, and none of the patients
had received radiotherapy or chemotherapy prior to surgery. For the
use of these clinical materials for research purposes, prior
consent from patients and approval from the Ethics Committee of The
Affiliated Hospital of Qingdao University were obtained. Informed
consent was provided to all study subjects. Patient anonymity has
been preserved. The pathological judgment was determined by two
senior pathologists with a double-blind study design. All specimens
were classified according to WHO (World Health Organization)
criteria.
Isolation of microRNA from HCC cells,
liver tissues and reverse transcription and qRT-PCR
Total RNA was extracted from the HCC tissues and
adjacent non-tumorous tissues by TRIzol reagent (Invitrogen). For
miRNA isolation and detection, reverse-transcribed complementary
DNA was synthesized using the PrimeScript RT reagent kit (Takara
Bio, Dalian, China) and quantitative real-time-PCR (qRT-PCR) was
performed using SYBR Premix Ex Taq (Takara) with the real-time PCR
system (Takara Bio). The PCR cycling conditions were as follows:
pre-denaturing at 95°C for 10 min, followed by 40 cycles of
denaturing at 95°C for 10 sec, annealing at 58°C for 20 sec and
extension at 72°C for 10 sec. Expression levels were normalized to
the endogenous snRNA U6 control. The relative expression ratio of
miR-429, in each paired tumor and non-tumorous tissue, was
calculated by the 2−ΔΔCT method. For the mRNA analysis,
cDNA was synthesized using reverse transcriptase kit (Takara Bio).
qRT-PCR was performed with SYBR Premix Ex Taq with the Takara SYBR
real-time PCR system. GAPDH was used as an internal control for
mRNA quantification. The relative expression ratio of mRNA was
calculated by the 2−ΔΔCT method. PCR reactions for each
gene were repeated three times. Independent experiments were
performed in triplicate.
Cell viability assay
Cell viability was examined by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
cell viability kit following the instructions of the manufacturer.
Cells were cultured in plastic 96-well plates under 200 µl of
growth medium and at an initial density of 10,000 cells/well. After
the treatment at the indicated time, 20 µl of 5 mg/ml MTT was
added. After 2 h, cells were lysed with 150 µl isopropanol/HCl
(0.05 M HCl in 100% isopropanol), and the optical density of each
well was determined at 450 nm on a Dynatech MR5000 microplate
reader (Dynatech Corp., Burlington, MA, USA). For each measurement,
both treatment and control, eight replicate wells were
recorded.
Transwell migration assays
The migration assays were performed using 24-well
Transwell® chambers (8 µm; Corning Incorporated,
Corning, NY, USA). For the migration assay, tumor cells were
resuspended in Dulbeccos modified Eagles medium (DMEM) and
2×105 cells were seeded into the upper chambers. Then,
0.5 ml DMEM containing 10% FBS was added to the bottom chambers.
Following a 24-h incubation, cells on the upper surface of the
membrane were scrubbed off, and the migrated cells were fixed with
95% ethanol, stained with 0.1% crystal violet and counted under a
light microscope (Olympus Corp., Tokyo, Japan).
Transient transfection with siTRAF6 or
pcDNA-TRAF6
SiTRAF6 were designed and synthesized by Shanghai
GenePharma, Co., Ltd. (Shanghai, China). The sequence of the
negative control (siCON) was also designed by Shanghai GenePharma.
The TRAF6-overexpressing plasmid vector (pcDNA-TRAF6) was
previously constructed and preserved in our laboratory. Twelve
hours prior to the transfection, the cells were plated in a 6-well
or a 96-well plate (Corning Incorporated). Lipofectamine 2000
reagent (Invitrogen) was then used to transfect siTRAF6 or
pcDNA-TRAF6 into the cells according to the manufacturers
protocol.
Western blot analysis
As previously described, target protein levels were
evaluated by western blot assays. Briefly, cells were lysed in 100
µl cold Triton lysis buffer (20 mM Tris pH 7.4, 2 mM PPiNa, 2 mM
EDTA pH 7.4, 137 mM NaCl, 25 mM β-glycerophosphate pH 7.4, 10%
glycerol, 1% Triton X-100) supplemented with 1 mM
Na3VO4, 0.5 mM DTT, 1 mM PMSF, and 1X
complete protease inhibitor cocktail (Roche) for 15 min at 4°C.
Lysates were spun at 14,000 rpm for a period of 10 min at 4°C.
Protein concentration was determined using the Bradford reagent
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Total cell
extracts (30 µg) were separated in 12% SDS-PAGE gels, transferred
to PVDF membranes (Millipore, Temecula, CA, USA) and probed with
antibodies against c-Myc (1:1,000 dilution; Abcam, Cambridge, UK),
total TAK1 (1:1,000 dilution; Abcam), phosphorylation of TAK1
(1:1,000 dilution; Abcam), TRAF6 (1:1,000 dilution; Abcam) or GAPDH
(1:2,000 dilution; Abcam). After incubation with the primary
antibodies, the membranes were washed with TBS/0.05% Tween-20 and
incubated with horseradish peroxidase-conjugated secondary
antibodies at room temperature for 1 h. Signals were detected using
enhanced chemiluminescence reagents (Pierce, Rockford, IL,
USA).
3-UTR luciferase reporter assays
To generate the 3-UTR luciferase reporter, the
partial sequence of the 3-UTR from TRAF6 was cloned downstream of
the firefly luciferase gene in the pGL3-control vector (Promega,
Madison, WI, USA). Mutation of the miR-429 target sites in the
3-UTR of TRAF6 was used as a control. pRL-TK containing
Renilla luciferase was co-transfected for data
normalization. For the luciferase reporter assays, 3×105
HEK293 cells were seeded in 35-mm culture plates, allowed to adhere
overnight, and then transfected with either 100 ng of empty
pGL3-control vector, 100 ng of the pGL3/TRAF6 construct, or 100 ng
of the pGL3/TRAF6-mut construct together with either 200 ng of
miR-CON or miR-429 using Lipofectamine 2000 (Invitrogen). Two days
later, the cells were harvested and assayed with the
Dual-luciferase assay (Promega) according to the manufacturers
instructions. Each treatment was performed in triplicate in three
independent experiments. The results are expressed as the relative
luciferase activity (Firefly LUC/Renilla LUC). After
luciferase activity was calculated for each miRNA, all were
normalized taking miR-CON data as 1.
Tumorigenicity assays in nude
mice
BALB/c athymic nude mice (male, 6-weeks old, 16–20
g) were purchased from Beijing HFK Bioscience, Co., Ltd. (Beijing,
China) and bred under pathogen-free conditions. All animal
experiments were approved by the Affiliated Hospital of Qingdao
University Institutional Animal Care and Use Committee. Briefly,
athymic nude mice were randomly divided into two groups, each
containing 6 mice. Each mouse injected the right flank with
5×106 LV-miR-429- or LV-miR-CON-infected SK-HEP1 cells
in 150 µl phosphate-buffered saline (PBS). Thereafter, tumor sizes
were measured every other day. When the sizes of the tumors were
above the limit described by the animal protocol approved by the
Affiliated Hospital of Qingdao University Medical College
Institutional Animal Care, the mice were sacrificed and the tumors
were removed. Tumors were fixed in 10% buffered formalin
(HistoChoice Tissue Fixative MB; Amresco, Solon, OH, USA) then
processed and embedded in paraffin. Immunohistochemistry was
performed on 5-µm-thick paraffin sections mounted on charged
slides. The sections were stained with H&E and immunostained
with Ki-67 to observe proliferation. The sections were also
immunostained to detect the expression of P65. For
immunohistochemistry (IHC) staining, endogenous peroxidase was
blocked using a 3% hydrogen peroxide solution. Primary antibodies
(P65 with 1:100 dilution, Ki-67 with 1:100 dilution) were incubated
overnight at 4°C in antibody diluent with background reducing
components (Beijing Zhongshan Jinqiao Biotechnology, Co., Ltd.,
Beijing, China). Secondary antibody, 1:250 HRP labeled
anti-mouse/rabbit (Abcam), incubation was performed at room
temperature for 30 min, and bound peroxidase was detected using the
ABC Peroxidase kit (Beijing Zhongshan Jinqiao Biotechnology) and
DAB (Beijing Zhongshan Jinqiao Biotechnology). All IHC slides were
counterstained with hematoxylin.
Statistical analysis
Data were analyzed using the two-tailed Students
t-test. A P<0.05 was considered statistically significant.
Results
Downregulation of miR-429 is
correlated with the progression of HCC
To measure the expression level of miR-429 in HCC,
quantitative real-time polymerase chain reaction (qRT-PCR) analysis
was performed using 25 pairs of snap-frozen human primary HCC and
corresponding adjacent tissue. As shown in Fig. 1A, miR-429 expression, in HCC
tissues, was significantly lower than that in pair-matched adjacent
tissue (P<0.01). These results suggested that miR-429
downregulation showed a positive correlation with the progression
of HCC. Next, we determined whether miR-429 expression was
similarly correlated in HCC cells lines. We compared miR-429
expression in five selected cell lines: HL7702 (human normal liver
cells), SK-HEP1, HepG2, SMMC7721 and HCCLM3. Indeed, miR-429 was
downregulated in the HCC cell lines compared with the normal liver
cells HL7702 (Fig 1B). Among these
HCC cell lines, miR-429 was upregulated in HCCLM3, which is used
for anti-miR-429 experiment in the following process. On the
contrary, miR-429, expressed at a low-level in the SK-HEP1, was
upregulated in the miR-429-overexpression experiment. Collectively,
the data indicated that miR-429 downregulation was significantly
associated with the proliferation of HCC.
Overexpression of miR-429 suppresses
HCC cell proliferation and migration
To investigate the effect of miR-429 on HCC cell
proliferation and motility, we successfully constructed a
recombinant lentiviral vector named LV-miR-429, LV-anti-miR-429
(miR-429 downregulation), and LV-miR-CON (and LV-miR-CON as
control) to infect HCCLM3 or SK-HEP1 cells. After successful
infection, miR-429 expression was confirmed by qRT-PCR (Fig. 2A). The results of the MTT assay
showed that overexpression of miR-429 significantly suppressed HCC
cell proliferation (Fig. 2B). Then,
cell motility was measured by wound-healing assay and Transwell
migration assays. The results showed that overexpression of miR-429
effectively suppressed HCC cell migration (Fig. 2C-F). These results revealed that
miR-429 suppressed HCC cell proliferation and motility.
TRAF6 is a direct target of
miR-429
To further elucidate the mechanism by which miR-429
affected cell proliferation and migration, we screened for
potential targets of miR-429 using three target prediction programs
with different algorithms: TargetScan, miRanda and PicTar. Based on
the representation of the miR-429 sites in their 3-UTRs, >100
mRNAs were predicted to be regulated by miR-429. Genes with
>2-fold changes in expression were considered of interest. Among
these candidates, the following six genes, TNF receptor associated
factor 6 (TRAF6), zinc finger E-box binding homeobox 2 (ZEB2),
WAS/WASL interacting protein family member 1 (WIPF1), neuregulin 1
(NRG1), heat shock protein family A (Hsp70) member 9 (HSPA9) and
leber congenital amaurosis 5 (LCA5) were involved in the cancer
proliferation and migration. TRAF6, a tumor promoter in human
tumorigenesis, was the most downregulated among all miR-429 target
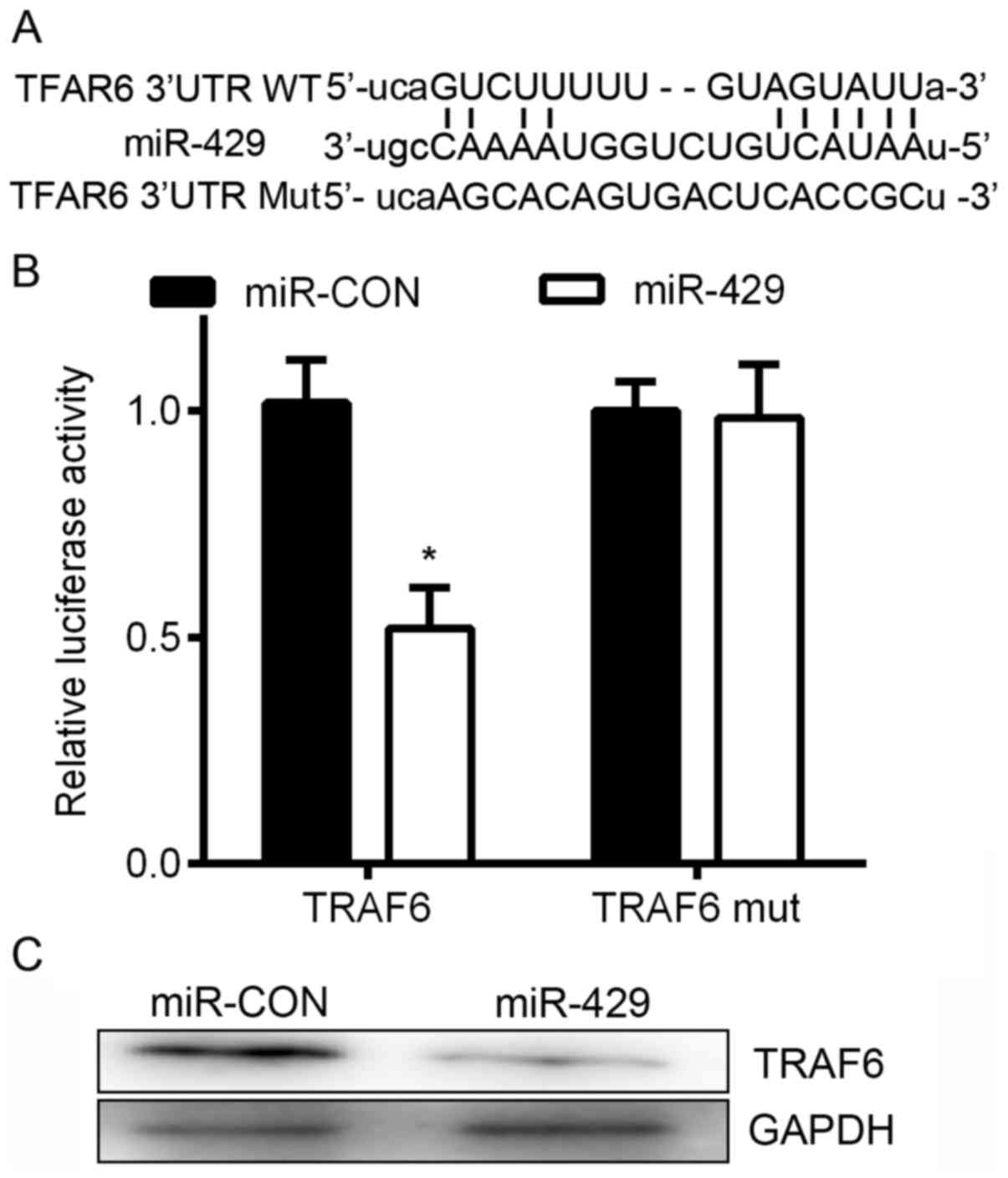
genes (Fig. 3A). To confirm this
finding, we subcloned the 3-UTR region of TRAF6 mRNA, including the
predicted miR-429 recognition site (wild-type, WT) or the mutated
site (mutant type, Mut) into luciferase reporter plasmids. A
Dual-luciferase reporter system was used. We found that the
overexpression of miR-429 significantly suppressed WT, but not Mut
3-UTR of TRAF6 (Fig. 3B). In
addition, the western blot results showed that the overexpression
of miR-429 significantly decreased the protein level of TRAF6
(Fig. 3C). Collectively, these
results suggest that miR-429 downregulates TRAF6 expression by
directly targeting its 3-UTR.
TRAF6 downregulation partially
attenuates the effect of miR-429
To further investigate whether downregulation of
TRAF6 could attenuate the effect of miR-429 on HCC cells, siCON or
siTRAF6 or LV-anti-miR-429 was transfected into HCCLM3 cells and
the effect was validated by western blot analysis (Fig. 4A). The results of the MTT (Fig. 4B), wound-healing and Transwell
migration assays (Fig. 4C-F) all
showed that TRAF6 downregulation could significantly attenuate the
effect of miR-429 on HCC cells. The data suggest that miR-429
inhibits HCC cell proliferation and motility, partially by
targeting TRAF6.
Overexpreesion of miR-429 inhibits
cell proliferation and NF-κB/TCF-4 signaling
To further study the role of miR-429 in inhibiting
HCC cancer cell proliferation, miR-429 was overexpressed in SK-HEP1
cells. We found that the protein expression of pTAK1
(phosphorylation TAK1) and c-Myc was significantly decreased in
cells infected with LV-miR-429 in comparison with those transfected
with the LV-miR-CON (Fig. 5A and
B). In contrast, co-transfection of LV-miR-429 and pCDNA-TRAF6
resulted in opposite effects on the levels of pTAK1 and c-Myc.
Analysis of NF-κB localization by immunofluorescence indicated that
the fraction of cells with nuclear P65 also decreased following
upregulation of miR-429 (Fig. 5C and
D). Furthermore, nuclear localization of P65 was increased in
response to pCDNA-TRAF6 transfection (Fig. 5C and D), suggesting that miR-429
upregulation is sufficient to decrease cell proliferation and NF-κB
signaling in at least a portion of HCC cancer cells.
miR-429 inhibits HCC tumor growth in
nude mice
Considering the important role of miR-429 in HCC, we
next used HCC xenograft models to further confirm the above
findings in vivo. Because miR-429 was significantly
downregulated in HCC, we successfully infected SK-HEP1 cells with
LV-miR-429 or LV-miR-CON. The LV-miR-429 or LV-miR-CON-infected
SK-HEP1 cells were then injected into the right flank of athymic
nude mice to establish subcutaneous HCC xenografts. Twenty-four
days after the injection, the tumors were removed and measured. The
tumor sizes in the LV-miR-429 group were much smaller than those in
the LV-miR-CON group (Fig. 6A and
B). To clarify the cellular mechanisms underlying
miR-429-mediated tumor growth, resected tissues from the
subcutaneous xenograft tumors were analyzed to verify the
expression of Ki-67 and P65. As shown in Fig. 6C and D, the LV-miR-429 group
displayed reduced Ki-67 and P65 expression in the tumor tissues.
The data provide strong evidence that miR-429 can inhibit HCC
growth in vivo.
Discussion
Accumulated studies have shown that miRNAs play a
crucial role in the process of tumor formation (14). It is well known that dysregulation
of miRNAs is frequently observed in multiple types of cancers and
plays fundamental roles in tumor initiation and progression. miRNAs
impact the dynamic balance between oncogenes and tumor suppressor
genes by degrading target genes, thereby, contributing to cancer
progression (15). In the present
study, we demonstrated that miR-429 was frequently downregulated in
HCC tissues in comparison to non-tumorous liver tissues. In
addition, its expression level is also decreased in HCC cell lines
compared with normal liver cells. However, the role of miR-429 in
HCC proliferation and migration remains unknown.
Previous studies have shown that the expression of
miR-429 is significantly downregulated in several types of cancers,
such as gastric (16), bladder
(17), breast cancer (18), glioma (19), colorectal carcinoma (20) and oral squamous cell carcinoma
(21). Ye and colleagues (18) found that overexpression of miR-429
in MDA-MB-231 cells remarkably suppressed invasion in vitro.
Additionally, Zhang and colleagues (16) found that miR-429 is significantly
downregulated in gastric cancer tissues in comparison to matched
non-tumorous tissues. Overexpression of miR-429 in gastric cancer
cells suppressed cell proliferation. Moreover, Sun and colleagues
(20) showed that miR-429 was
significantly downregulated in colorectal carcinoma (CRC) tissues
and cell lines. The results reveal that miR-429 inhibited the
proliferation and growth of CRC cells both in vitro and
in vivo, suggesting that miR-429 could play a role in CRC
tumorigenesis.
miR-429 significantly decreased the migration and
proliferation of HCC cells in vitro, and reduced their
capacity to develop HCC tumors in vivo. However, some
investigators observed an opposite pattern of miR-429 expression in
other cancers. For example, Lang et al (22) found that miR-429 are often
upregulated in non-small cell lung cancer (NSCLC) in comparison to
normal lung tissues, and its expression level is also increased in
NSCLC cell lines in comparison to normal lung cells. High
expression levels of miR-429 in A549 NSCLC cells significantly
promoted cell proliferation, migration, and invasion, whereas
inhibition of miR-429 inhibits these effects in vitro
(22). It has been reported that
the overexpression of miR-429 leads to a decrease in PTEN, RASSF8
and TIMP2 expression, whereas an adverse effect is observed when
miR-429 is downregulated in NSCLC. Moreover, miR-429 has been found
to be a potential target for NSCLC therapy. It was recently
reported that ectopic expression of miR-429 markedly induced the
expression of MMP2/7/9 and enhanced HCC migration and invasion
in vitro and in vivo via regulating the classic Wnt
pathway (23). Epigenetic
modification of miR-429 can manipulate liver tumour-initiating
cells by targeting the Rb binding protein 4 (RBBP4)/E2F1/OCT4 axis
(24). However, miR-429 directly
targeted NOTCH1 and reduced both mRNA and protein levels of NOTCH1
which stimulated proliferation and suppressed apoptosis in HCC
cells in HBV-related HCC (25).
Therefore, we conducted these experiments in order to answer this
question regarding the relationship between miR-429 expression and
HCC.
Through the use of TargetScan, miRanda and PicTar,
we were able to predict that TRAF6 was a probable target of miR-429
in the 3-UTR of TRAF6. We demonstrated that miR-429 overexpression
resulted in the downregulation of TRAF6 at the protein level,
whereas functional inhibition of miR-429 led to the upregulation of
TRAF6. These results strongly suggest that TRAF6 is regulated by
miR-429 in HCC. Moreover, a Dual-luciferase reporter assay
identified TRAF6 as a direct target of miR-429.
Tumor necrosis factor receptor-associated factors
(TRAFs) are a family of adaptor proteins that couple tumor necrosis
factor receptor families to signaling pathways (26). As a key activator of nuclear
factor-κB (NF-κB), TNF receptor-associated factor 6 (TRAF6) is
known to transduce activating signals from the TNFR or Toll-like
receptor (TLR)/IL-1 receptor family to NF-κB (27). At the molecular level, TRAF6
functions as an E3 ubiquitin ligase that activates IκB kinase
(IKK), resulting in degradation of IκB, as well as nuclear
translocation and activation of NF-κB as a physiologic
response.TRAF6 has also been intermittently investigated in various
cancer cells (28,29). Activation of some signaling
mechanisms, including the NF-κB pathway, has been suggested to be
involved in TRAF6-mediated oncogenesis (30). The generation of TRAF6 is essential
for the recruitment of the downstream kinase TGFβ-activated kinase
1 (TAK1), which forms a complex with the ubiquitin-binding
TAK1-binding proteins, TAB1, −2 and −3 (31). Subsequently, TAK1 activates the
NF-κB pathway. In the present study, we provide solid evidence
demonstrating a strong relationship between TRAF6 expression and
HCC oncogenicity both in vitro and in vivo.
Previously, it has also been shown that TRAF6 is able to modulate
the degradation of TAK1 (32),
which could impair the downstream effectors for pathogenesis.
Therefore, a potential linkage between miR-429 and the
phosphorylation of TAK1 was investigated. In HCC cells, TRAF6 gene
silencing by short hairpin RNA led to a significant increase in
NF-κB-dependent gene activity.
NF-κB is frequently activated in various types of
tumors and promotes cancer development (33). Therefore, miRNAs that possess the
NF-κB inhibitory activity may provide novel targets for anticancer
therapy. Our data reveal a significant inhibitory role of miR-429
on NF-κB signaling and a reduced nuclear accumulation of NF-κB p65.
Therefore, our results suggest that miR-429 downregulation is a
novel mechanism that contributes to the abnormal activation of the
NF-κB pathway in HCC cells. We demonstrated that miR-429
inactivated NF-κB signaling in order to inhibit migration and
decrease motility of HCC cells. Previously, the level of TRAF6 also
revealed a positive correlation with P65 expression (34). Therefore, the present study revealed
a novel mechanism by which miR-429 regulates NF-κB signaling and
contributes to cancer proliferation in vitro and in
vivo. It is exciting to find that a single miRNA may suppress
tumor growth via NF-κB signaling, which makes miR-429 a promising
anticancer target in HCC.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundations of China (grant nos. 81372632
and 81402579), the Special Funds of Post Doctoral Innovation
Projects of Shandong Province of China (grant no. 201303063), and
the Application of Post Doctoral funds of Qingdao city, Shandong
Province of China (grant no. 20130118).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marquardt JU and Thorgeirsson SS:
SnapShot: Hepatocellular carcinoma. Cancer Cell. 25:550.e5512014.
View Article : Google Scholar
|
|
3
|
Agrawal S, Agarwal S, Arnason T, Saini S
and Belghiti J: Management of hepatocellular adenoma: Recent
advances. Clin Gastroenterol Hepatol. 13:1221–1230. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mehlen P and Puisieux A: Metastasis: A
question of life or death. Nat Rev Cancer. 6:449–458. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shenouda SK and Alahari SK: MicroRNA
function in cancer: Oncogene or a tumor suppressor? Cancer
Metastasis Rev. 28:369–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oishi N, Yamashita T and Kaneko S:
Molecular biology of liver cancer stem cells. Liver Cancer.
3:71–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bandiera S, Pfeffer S, Baumert TF and
Zeisel MB: miR-122 - a key factor and therapeutic target in liver
disease. J Hepatol. 62:448–457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kota J, Chivukula RR, ODonnell KA, Wentzel
EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M,
Clark KR, et al: Therapeutic microRNA delivery suppresses
tumorigenesis in a murine liver cancer model. Cell. 137:1005–1017.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee CH, Kim JH and Lee SW: The role of
microRNAs in hepatitis C virus replication and related liver
diseases. J Microbiol. 52:445–451. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yuan Q, Loya K, Rani B, Möbus S,
Balakrishnan A, Lamle J, Cathomen T, Vogel A, Manns MP, Ott M, et
al: MicroRNA-221 overexpression accelerates hepatocyte
proliferation during liver regeneration. Hepatology. 57:299–310.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang JY, Zhang K, Chen DQ, Chen J, Feng
B, Song H, Chen Y, Zhu Z, Lu L, De W, et al: MicroRNA-451:
Epithelial-mesenchymal transition inhibitor and prognostic
biomarker of hepatocelluar carcinoma. Oncotarget. 6:18613–18630.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng Z, Liu J, Yang Z, Wu L, Xie H, Jiang
C, Lin B, Chen T, Xing C, Liu Z, et al: MicroRNA-452 promotes
stem-like cells of hepatocellular carcinoma by inhibiting Sox7
involving Wnt/β-catenin signaling pathway. Oncotarget.
7:28000–28012. 2016.PubMed/NCBI
|
|
13
|
Tan G, Wu L, Tan J, Zhang B, Tai WC, Xiong
S, Chen W, Yang J and Li H: MiR-1180 promotes apoptotic resistance
to human hepatocellular carcinoma via activation of NF-κB signaling
pathway. Sci Rep. 6:223282016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hall DP, Cost NG, Hegde S, Kellner E,
Mikhaylova O, Stratton Y, Ehmer B, Abplanalp WA, Pandey R, Biesiada
J, et al: TRPM3 and miR-204 establish a regulatory circuit that
controls oncogenic autophagy in clear cell renal cell carcinoma.
Cancer Cell. 26:738–753. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheng CJ, Bahal R, Babar IA, Pincus Z,
Barrera F, Liu C, Svoronos A, Braddock DT, Glazer PM, Engelman DM,
et al: MicroRNA silencing for cancer therapy targeted to the tumour
microenvironment. Nature. 518:107–110. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang M, Dong BB, Lu M, Zheng MJ, Chen H,
Ding JZ, Xu AM and Xu YH: miR-429 functions as a tumor suppressor
by targeting FSCN1 in gastric cancer cells. Onco Targets Ther.
9:1123–1133. 2016.PubMed/NCBI
|
|
17
|
Wu CL, Ho JY, Chou SC and Yu DS: MiR-429
reverses epithelial-mesenchymal transition by restoring E-cadherin
expression in bladder cancer. Oncotarget. 7:26593–26603.
2016.PubMed/NCBI
|
|
18
|
Ye ZB, Ma G, Zhao YH, Xiao Y, Zhan Y, Jing
C, Gao K, Liu ZH and Yu SJ: miR-429 inhibits migration and invasion
of breast cancer cells in vitro. Int J Oncol. 46:531–538.
2015.PubMed/NCBI
|
|
19
|
Chen W, Zhang B, Guo W, Gao L, Shi L, Li
H, Lu S, Liu Y and Li X: miR-429 inhibits glioma invasion through
BMK1 suppression. J Neurooncol. 125:43–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun Y, Shen S, Liu X, Tang H, Wang Z, Yu
Z, Li X and Wu M: MiR-429 inhibits cells growth and invasion and
regulates EMT-related marker genes by targeting Onecut2 in
colorectal carcinoma. Mol Cell Biochem. 390:19–30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lei W, Liu YE, Zheng Y and Qu L: MiR-429
inhibits oral squamous cell carcinoma growth by targeting ZEB1. Med
Sci Monit. 21:383–389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lang Y, Xu S, Ma J, Wu J, Jin S, Cao S and
Yu Y: MicroRNA-429 induces tumorigenesis of human non-small cell
lung cancer cells and targets multiple tumor suppressor genes.
Biochem Biophys Res Commun. 450:154–159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang J, Li L, Huang W, Sui C, Yang Y, Lin
X, Hou G, Chen X, Fu J, Yuan S, et al: MiR-429 increases the
metastatic capability of HCC via regulating classic Wnt pathway
rather than epithelial-mesenchymal transition. Cancer Lett.
364:33–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li L, Tang J, Zhang B, Yang W, LiuGao M,
Wang R, Tan Y, Fan J, Chang Y, Fu J, et al: Epigenetic modification
of MiR-429 promotes liver tumour-initiating cell properties by
targeting Rb binding protein 4. Gut. 64:156–167. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao H and Liu C: miR-429 represses cell
proliferation and induces apoptosis in HBV-related HCC. Biomed
Pharmacother. 68:943–949. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bradley JR and Pober JS: Tumor necrosis
factor receptor-associated factors (TRAFs). Oncogene. 20:6482–6491.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chung JY, Park YC, Ye H and Wu H: All
TRAFs are not created equal: Common and distinct molecular
mechanisms of TRAF-mediated signal transduction. J Cell Sci.
115:679–688. 2002.PubMed/NCBI
|
|
28
|
Peng Z, Shuangzhu Y, Yongjie J, Xinjun Z
and Ying L: TNF receptor-associated factor 6 regulates
proliferation, apoptosis, and invasion of glioma cells. Mol Cell
Biochem. 377:87–96. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
He Z, Huang C, Lin G and Ye Y:
siRNA-induced TRAF6 knockdown promotes the apoptosis and inhibits
the invasion of human lung cancer SPC-A1 cells. Oncol Rep.
35:1933–1940. 2016.PubMed/NCBI
|
|
30
|
Lin D, Zhang M, Zhang MX, Ren Y, Jin J,
Zhao Q, Pan Z, Wu M, Shu HB, Dong C, et al: Induction of USP25 by
viral infection promotes innate antiviral responses by mediating
the stabilization of TRAF3 and TRAF6. Proc Natl Acad Sci USA.
112:11324–11329. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xiao F, Wang H, Fu X, Li Y and Wu Z: TRAF6
promotes myogenic differentiation via the TAK1/p38
mitogen-activated protein kinase and Akt pathways. PLoS One.
7:e340812012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Walsh MC, Lee J and Choi Y: Tumor necrosis
factor receptor- associated factor 6 (TRAF6) regulation of
development, function, and homeostasis of the immune system.
Immunol Rev. 266:72–92. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Perkins ND: The diverse and complex roles
of NF-κB subunits in cancer. Nat Rev Cancer. 12:121–132.
2012.PubMed/NCBI
|
|
34
|
Starczynowski DT, Lockwood WW, Deléhouzée
S, Chari R, Wegrzyn J, Fuller M, Tsao MS, Lam S, Gazdar AF, Lam WL,
et al: TRAF6 is an amplified oncogene bridging the RAS and NF-κB
pathways in human lung cancer. J Clin Invest. 121:4095–4105. 2011.
View Article : Google Scholar : PubMed/NCBI
|