Introduction
Squamous cell carcinoma (SCC) accounts for most oral
cancers (1). Despite improvements
in diagnosis and treatment, oral SCC (OSCC) is still associated
with a poor prognosis and high mortality rate (2,3). One
property of OSCC is that it is propagated by progressive local
invasion (4,5) and lymphatic metastasis, correlating
with the clinical stage (6). Thus,
a better understanding of the mechanisms of invasion is important
for improving the treatments for OSCC. Aspects of tumour invasion
include cell migration, tumour-stroma interactions at the invasive
front and the influence of external stimuli, including growth
factors (7–11).
Receptor tyrosine kinases (RTKs) mediate many
fundamental effects in cells, including regulating migration
(12). An example of RTK-activating
growth factor is the epidermal growth factor (EGF) family, members
of which act on the EGF receptor (EGFR) (13). Overexpression of EGFR has been
detected in ~90% of head and neck squamous cell carcinomas and
correlates with the clinical stage (14) and EGFR is a factor influencing poor
prognosis (15). The EGFR
inhibitors, including the monoclonal antibody cetuximab and
small-molecule inhibitors (e.g., gefitinib, erlotinib and
afatinib), have been used in the clinic to block signalling
downstream of EGFR via ligand binding and ATP-binding sites of
EGFR, respectively (16,17).
Previously, we showed that EGFR inhibitors
suppressed cell migration in OSCC SAS cells but not HSC4 cells
(18,19). Another RTK involved in the
regulation of motility is c-Met, activated by hepatocyte growth
factor (HGF, also known as scatter factor) (20,21).
c-Met is expressed in various cell types including epithelial and
vascular endothelial cells, and its ligand HGF is released from
stromal and some tumour cells (22,23).
In migrating cells, filopodia and lamellipodia
formation, by remodelling of the actin cytoskeleton, is observed at
the leading edge of the moving side (24). Activation of the Rho family members
Rac1 and Cdc42 via the PI3K/Akt or MEK/ERK pathway promotes the
formation of lamellipodia and filopodia in prostate and breast
cancers (25–27). c-Met/PI3K/Akt signalling plays a
role in HGF-mediated lamellipodia formation and motility in lung
endothelial cells (28).
Furthermore, lamellipodin, a protein necessary for the formation of
lamellipodia, has been reported to be important in cell migration
via interactions with the Ena/VASP or Scar/WAVE complex (29,30).
Although Wnt signalling promotes formation of pseudopodia by the
stimulating Cdc42 and RhoA in OSCC (31), other protein levels and signalling
pathways necessary for the formation of pseudopodia in OSCC have
yet to be determined.
In the present study, we examined the importance of
EGFR and the c-Met signalling pathway in cell migration via
filopodia and lamellipodia formation using OSCC cell lines.
Materials and methods
Cell culture and reagents
Three OSCC cell lines, HSC4, SAS and Ca9-22, were
purchased from the RIKEN BioResource Center (Ibaraki, Japan). Cells
were cultured in Dulbecco's modified Eagle's medium (DMEM),
supplemented with 10% (v/v) fetal bovine serum (FBS) at 37°C in a
humidified atmosphere of 5% CO2. DMEM and FBS were
purchased from Gibco (Life Technologies, Tokyo, Japan). The
antibodies used included anti-c-Met (Cell Signaling Technology,
Tokyo, Japan), anti-phospho-c-Met (Tyr1234/1235, Cell Signaling
Technology), anti-lamellipodin (Cell Signaling Technology),
anti-cortactin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and
anti-α-tubulin (Sigma-Aldrich, Tokyo, Japan). Acti-stain™ 488 was
purchased from Cytoskeleton, Inc. (Denver, CO, USA). Cetuximab
(Erbitux®) was purchased from Merck Serono, Co., Ltd.
(Tokyo, Japan). SU11274 was from Sigma-Aldrich and AG1478 from
Calbiochem (Merk Millipore, Tokyo Japan).
Scratch wound healing assay
Cell migration was determined using a scratch wound
healing assay, as described elsewhere (18,19)
with slight modifications. Briefly, semi-confluent cells in 12-well
plates were treated with 10 µg/ml mitomycin C for 4 h to block
proliferation. The cells were subsequently wounded using a sterile
200-µl pipette tip to generate a cell-free gap, ~0.3 mm in width.
Cells were then washed with phosphate-buffered saline (PBS) and
photographed to record the wound width at 0 h. One group of cells
was cultured in DMEM with 10% FBS as a control, while the other
groups were treated with various concentrations of cetuximab,
AG1478, SU11274, EGF or HGF. After incubation, photographs were
taken to evaluate migration.
Determination of lamellipodia
Cultured cells were fixed in 3.5% (w/v)
paraformaldehyde, permeabilized in 0.2% (v/v) Triton X-100 and
blocked in 2% (w/v) bovine serum albumin (BSA). The cells were
incubated with anti-cortactin antibody at 4°C overnight, followed
by Alexa Fluor 594-conjugated IgG (Thermo Fisher Scientific,
Yokohama, Japan) as the secondary antibody and Acti-stain 488
phalloidin (Cytoskeleton) for actin-fiber staining. After
incubation, SlowFade gold antifade reagent with
4′,6-diamidino-2-phenylindole (Invitrogen/Life Technologies) was
added to the cells. The specimens were observed by fluorescence
microscopy (Olympus IX73; Olympus, Tokyo, Japan). We determined
lamellipodia formation by evaluating fluorescent actin fibers and
cortactin co-localization at the cell periphery.
Western blotting
Cells were washed with PBS and then lysed in RIPA
buffer consisting of 150 mM NaCl, 10 mM Tris-HCl, pH 8.0, 1% (v/v)
Nonidet P-40, 0.5% (w/v) deoxycholic acid, 0.1% (w/v) SDS and 5 mM
EDTA with 1X Halt™ Protease Inhibitor Cocktail (Thermo Fisher
Scientific) and 1X Halt™ Protein Phosphatase inhibitor (Thermo
Fisher Scientific). The protein concentrations of the lysates were
determined using a BCA™ protein assay kit (Thermo Fisher
Scientific) and equal amounts of protein were subjected to
SDS-polyacrylamide gel electrophoresis. Separated proteins were
transferred electrophoretically to Clear Trans PVDF membranes
(Wako, Tokyo, Japan). Non-specific binding was blocked by
incubation with 5% (w/v) BSA in TBS/Tween-20 (TBS-T) for 1 h at
room temperature. Membranes were probed with antibodies in TBS-T
overnight at 4°C and then incubated with HRP-conjugated secondary
antibodies. Antibody-antigen complexes were detected using ECL Plus
Western blotting detection reagent (GE Healthcare, Little Chalfont,
England).
Statistical analyses
Statistical analyses were performed using the
one-way analysis of variance (ANOVA). Statistical significance
(*P<0.05, **P<0.01 and ***P<0.001) was evaluated using
unpaired Student's t-test to assess the differences between the
treated and the control samples.
Results
The migration potency of HSC4 cells is
induced by serum stimulation and HGF/c-Met signalling
Previously, we showed that the EGFR pathway promoted
migration of the OSCC cell line SAS but not the OSCC cell line
HSC4. The migration potency of HSC4 cells was induced by
stimulation of an unknown factor in serum other than EGF (19).
To assess whether the migration of other OSCC cells
is regulated by the EGFR pathway, we investigated the influence of
EGF and EGFR inhibitors on the migration of the gingival cancer
cell line Ca9-22. The migration activity of Ca9-22 cells, as well
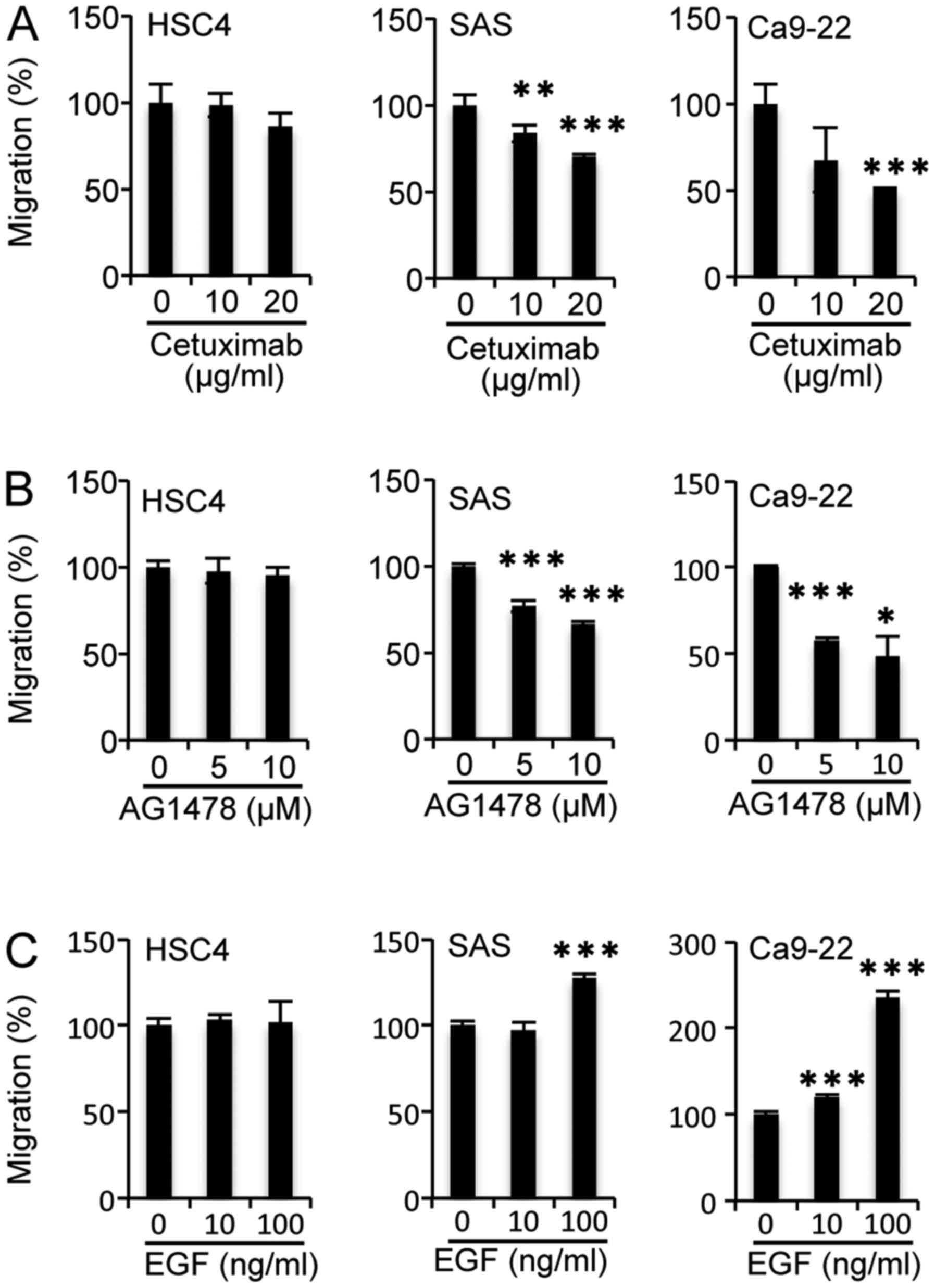
as SAS cells, was decreased significantly by cetuximab treatment at
20 µg/ml (Fig. 1A) and AG1478
treatment at 5 µM (Fig. 1B) but was
enhanced significantly by EGF addition at 100 ng/ml (Fig. 1C). Additionally, migration of HSC4
cells was unaffected by the addition of EGFR inhibitors or EGF
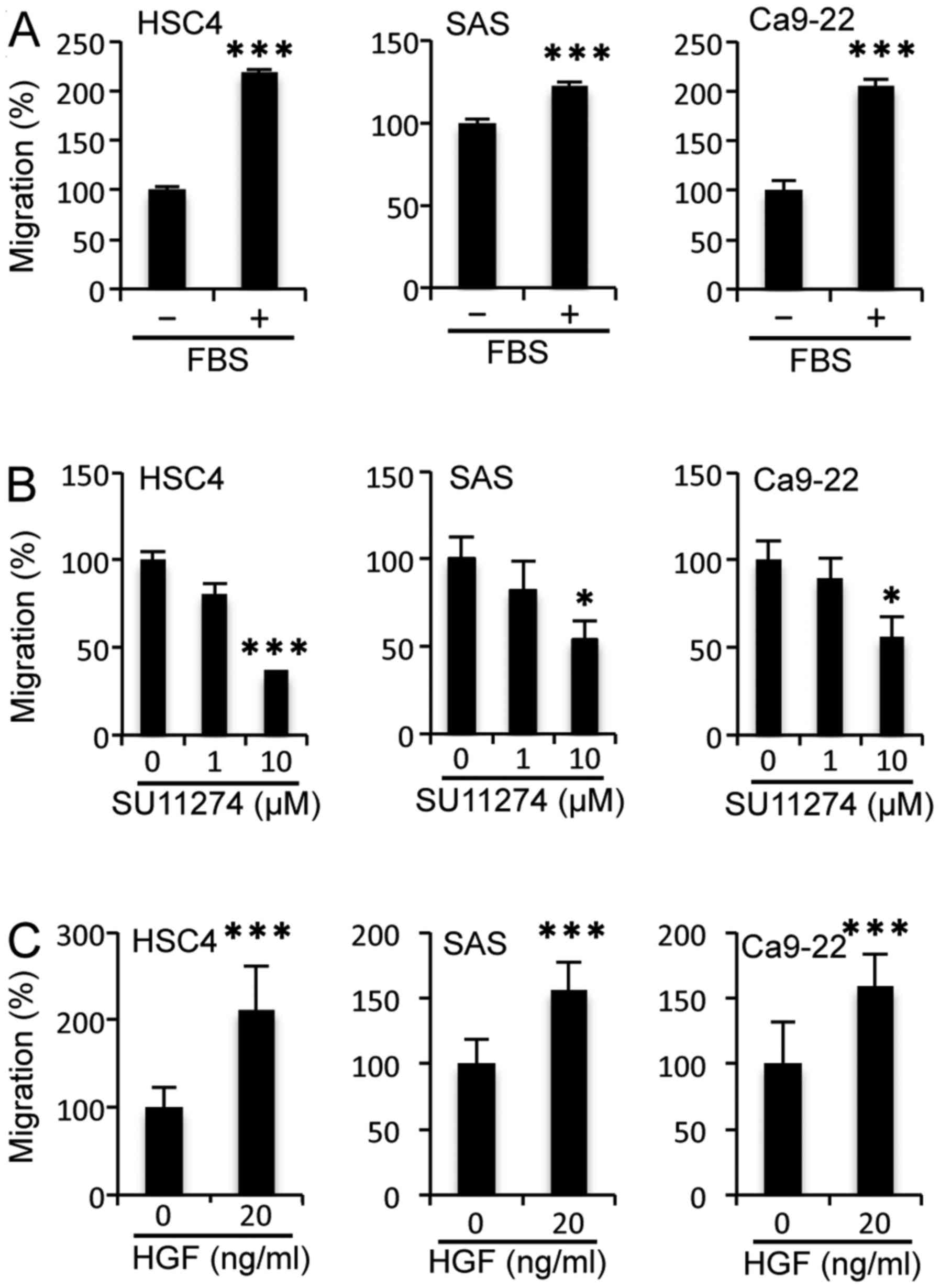
(Fig. 1). However, the addition of
serum induced the migration of HSC4 cells significantly, as it did
with SAS and Ca9-22 cells (Fig.
2A). These results indicate that cell migration was induced by
(a) serum component(s) other than EGF among the OSCC cell lines
examined.
Next, we investigated the effects of c-Met on cell
migration potency using a scratch wound healing assay in OSCC cell
lines, because c-Met is an RTK involved in cell migration along
with EGFR (20,32). When cells were treated with the
c-Met inhibitor SU11274 at 10 µM, the migratory potency of all cell
lines was reduced significantly (Fig.
2B). Moreover, the migratory activity of all cell lines was
enhanced significantly by addition of the c-Met ligand HGF at 20
ng/ml (Fig. 2C). These results
suggest that the migration of HSC4 cells is regulated by HGF/c-Met
signalling but not EGF/EGFR signalling. Furthermore, both EGF/EGFR
and HGF/c-Met signalling play a role in the migration of SAS and
Ca9-22 cells.
Filopodia and lamellipodia formation
is induced by EGFR signalling in OSCC SAS cells but not HSC4
cells
Cell migration is controlled by dynamic remodelling
of the actin cytoskeleton. In this process, the formation of
pseudopodia, including filopodia and lamellipodia, consisting of
actin fibres at the leading edge of the cells plays a pivotal role
(33). To understand the
relationship between the formation of filopodia and lamellipodia
and EGFR signalling in OSCC cells, we examined the effects of EGFR
inhibitor AG1478 and EGF treatment on filopodia and lamellipodia
formation in migrating cells facing the scratch wound. Filopodia
appeared in the untreated control and AG1478-treated HSC4 and SAS
cells by 0.5 h. Cells with filopodia increased in both cell lines
for over 1 h. After 12 h, some filopodia formation was observed in
almost all control and AG1478-treated HSC4 cells (Fig. 3A). However, although numbers of the
cells with filopodia increased after 12 h in SAS control cells, the
proportion of cells with filopodia was ~50% after 12 h of AG1478
treatment compared with the control (Fig. 3A).
We next examined the effects of EGF on filopodia
formation. In HSC4 and SAS cells, filopodia had formed in ~15–20%
by 0.5 h and in majority by 12 h regardless of EGF treatment
(Fig. 3B). These results showed
that EGFR signalling was necessary for filopodia formation in SAS
cells but not in HSC4 cells and that addition of EGF had no
apparent effect on filopodia formation.
Approximately 40% of untreated HSC4 and 80% of
untreated SAS cells formed lamellipodia by 12 h. The ratio of
lamellipodia formed in SAS cells treated with AG1478 was
significantly lower than that in control SAS cells after 6 h.
However, lamellipodia formation was not affected in AG1478 treated
HSC4 cells at 12 h (Fig. 3C). EGF
increased the number of SAS cells with lamellipodia, compared with
the control and had not apparent effect on HSC4 cells (Fig. 3D). These results suggest that the
EGFR pathway is involved in filopodia and lamellipodia formation in
SAS cells but not in HSC4 cells.
Filopodia and lamellipodia formation
is induced by HGF/c-Met signalling in HSC4 cells and SAS cells
We evaluated the effects of HGF/c-Met on filopodia
and lamellipodia formation in controlling migration potency in HSC4
and SAS cells. Filopodia formation in HSC4 and SAS cells treated
with SU11274 did not differ from that in the untreated cells after
0.5 and 1 h of culture. However, filopodia formation was reduced
significantly compared with untreated cells after 12 h of SU11274
treatment (Fig. 4A).
Because filopodia formation in HSC4 and SAS cells
was decreased by inhibition of c-Met signalling, we next examined
the effects of HGF, the ligand of c-Met. Filopodia-forming HSC4 and
SAS cells were increased after 1 h of HGF treatment compared with
the serum-free medium (Fig. 4B).
Additionally, the ratios of HSC4 and SAS cells with lamellipodia
was reduced significantly by SU11274 treatment for 6–12 h compared
with untreatment cells (Fig. 4C).
Furthermore, the percentages of lamellipodia-forming HSC4 and SAS
cells after HGF treatment did no differ from those of untreated
cells after 6 h but were increased after 12 h (Fig. 4D). These results suggest that
HGF/c-Met signalling is important for the formation of filopodia
and lamellipodia in HSC4 and SAS cells, and that EGFR signalling
plays an important role in filopodia and lamellipodia formation in
SAS cells.
Lamellipodin is regulated by HGF/c-Met
signalling but not by EGF/EGFR signalling, although c-Met
phosphorylation is regulated by EGFR signalling in HSC4 cells
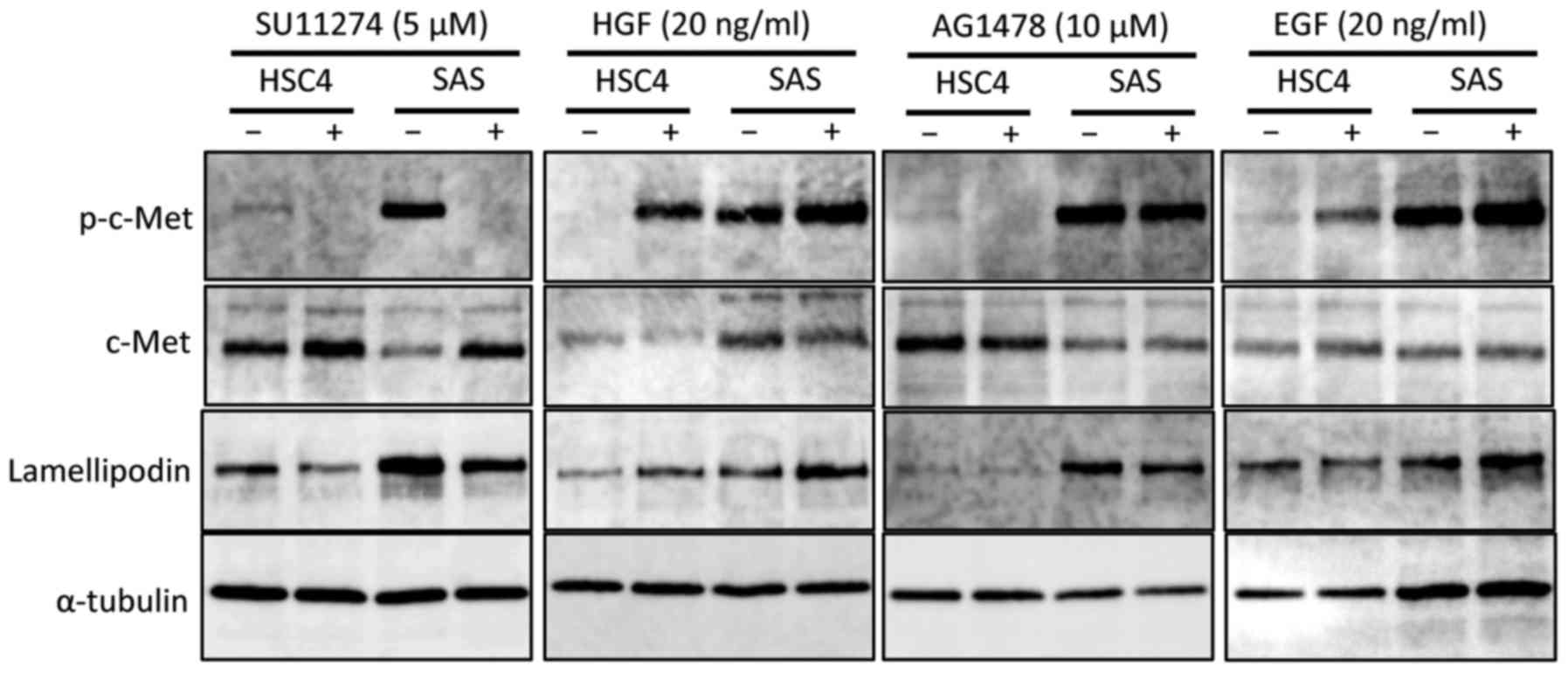
To further elucidate the role of HGF/c-Met and
EGF/EGFR signalling involved in the formation of lamellipodia in
the OSCC cell lines examined, we focused on the lamellipodin
protein, which is related to the formation of lamellipodia at the
leading edge, together with Ena/VASP proteins (34). We found that when the
phosphorylation level of c-Met was decreased by SU11274 treatment,
the lamellipodin level was also decreased in both HSC4 and SAS
cells. Lamellipodin levels and c-Met phosphorylation were both
upregulated by HGF stimulation in both HSC4 and SAS cells.
Furthermore, inhibition of EGFR signalling by AG1478 decreased
c-Met phosphorylation and lamellipodin levels in SAS cells.
However, although the phosphorylation level of c-Met was reduced by
AG1478, the level of lamellipodin was not changed in HSC4 cells.
Additionally, the levels of c-Met phosphorylation and lamellipodin
protein were increased by EGF stimulation in SAS cells. In
contrast, although c-Met phosphorylation was increased by EGF
stimulation, lamellipodin was unaffected in HSC4 cells.
These results suggest that EGFR signalling increases
the level of lamellipodin protein via a process involving of c-Met,
and that this promotes the formation of lamellipodia in SAS cells.
EGFR signalling could lead to phosphorylation of the c-Met, but it
does not affect the level of lamellipodin in HSC4 cells (Fig. 5).
Discussion
It has been reported that EGFR and c-Met are
involved in cell migration (20,35–38).
In the present study, we showed that migration of the OSCC cell
lines SAS and Ca9-22 was regulated by both the EGFR and c-Met
signalling pathways. In contrast, migration of OSCC HSC4 cells
involves c-Met activation but not the EGFR pathway. Indeed, HSC4
cell migration was resistant to EGFR inhibitor and sensitive to a
c-Met inhibitor. The mechanisms of EGFR inhibitor resistance have
been classified into two categories: developing of a secondary
mutation in EGFR and bypassing or activation of an alternative
pathway (39). We demonstrated
previously that the proliferation of HSC4 cells is sensitive to an
EGFR inhibitor (18). Treatment
with the EGFR inhibitor AG1478 decreased phosphorylation levels of
EGFR, AKT and ERK, as reported previously. These findings indicated
that the EGFR pathway plays a distinct role from that of the c-Met
pathway in HSC4 cells. c-Met played an important role in the
migration of all OSCC cell lines examined, suggesting that c-Met
may be an appropriate therapeutic target for invasion and
metastasis.
In the present study, we showed that the HGF/c-Met
pathway plays an important role in the formation of lamellipodia
and filopodia, as well as in the migration of OSCC cells. These
results are consistent with previous reports that HGF/c-Met
signalling promoted cell migration through lamellipodia and
filopodia formation in lung endothelial cells (28) and in some normal cells (30). Thus, it is possible that
lamellipodia and filopodia formation is regulated by c-Met
signalling, thereby promoting the migration of OSCC cells. ERK and
PI3K/Akt serve as downstream effectors of c-Met signalling
(28). However, the molecules
downstream of c-Met/ERK and c-Met/PI3K/Akt signalling that directly
regulate filopodia and lamellipodia formation remain unknown. In
this context, we showed that c-Met signalling was involved in the
regulation of lamellipodin protein levels. Promotion of cell
migration potency via the c-Met pathway is possibly regulated by
increasing the level of lamellipodin, because upregulation of
lamellipodin protein markedly promoted cell migration (30).
In this study, filopodia formation in SAS was
inhibited by an EGFR inhibitor, but was not promoted by EGF. These
data suggest that filopodia formation is regulated by
ligand-independent EGFR signalling or by EGFR ligands other than
EGF. Ligand-independent activation of EGFR, including Src-mediated
integrins (40,41) and G-protein coupled receptors
(42) has been reported. However,
there is no evidence yet that ligand-independent EGFR signalling
promotes filopodia formation. In contrast, heparin-binding EGF-like
growth factor (HB-EGF), an EGFR ligand, regulates invadopodia
(43) and invasion (44) via EGFR activation in some cancers.
Thus, the HB-EGF/EGFR pathway is likely involved in regulating
filopodia formation in SAS cells.
We found that the phosphorylation levels of c-Met
were increased by EGF and decreased by AG1478 in HSC4 and SAS
cells. These results suggest that EGFR activation by EGF results in
transactivation of c-Met. A possible mechanism for this
transactivation is that EGF/EGFR signalling upregulates HGF
production, thereby releasing HGF, which stimulates c-Met
phosphorylation. Ligand-activated c-Met via EGFR activation would
be expected to promote cell migration. EGF/EGFR signalling affected
migration in SAS cells but not HSC4 cells. Thus, the
EGF/EGFR/HGF/c-Met axis may play a role in the migration of SAS
cells. Another possibility is that lateral signalling from EGFR to
c-Met occurs through a certain mediator, for example, c-Src
(45). Such lateral signalling from
the EGFR/c-Src/c-Met axis may promote lamellipodia formation and
cell migration through lamellipodin upregulation in HSC4 and SAS
cells, although further investigation of this is needed.
In conclusion, we showed that the HGF/c-Met and
EGF/EGFR pathways increased the level of lamellipodin protein,
thereby inducing cell migration via lamellipodia formation in OSCC
cells. Further investigations of downstream effectors in c-Met
signalling will be useful for identifying potential new therapeutic
targets for OSCC patients.
Acknowledgements
The present study was edited by Textcheck English
consultants. This research was supported by the Osaka University
(M.N., no. 1508000001) and the Osaka Dental University (Y.O., no
217006). H.Y. is funded by the Osaka Dental University Research
Funds (no.15-07).
Glossary
Abbreviations
Abbreviations:
|
HGF
|
hepatocyte growth factor
|
|
SCC
|
squamous cell carcinoma
|
|
OSCC
|
oral SCC
|
|
HNSCC
|
head and neck SCC
|
|
EGF
|
epidermal growth factor
|
|
EGFR
|
EGF receptor
|
|
RTK
|
receptor tyrosine kinase
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
PBS
|
phosphate-buffered saline
|
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xi S and Grandis JR: Gene therapy for the
treatment of oral squamous cell carcinoma. J Dent Res. 82:11–16.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Magrath I and Litvak J: Cancer in
developing countries: Opportunity and challenge. J Natl Cancer
Inst. 85:862–874. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kramer RH, Shen X and Zhou H: Tumor cell
invasion and survival in head and neck cancer. Cancer Metastasis
Rev. 24:35–45. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ziober AF, Falls EM and Ziober BL: The
extracellular matrix in oral squamous cell carcinoma: Friend or
foe? Head Neck. 28:740–749. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Berenson JR, Yang J and Mickel RA:
Frequent amplification of the bcl-1 locus in head and neck squamous
cell carcinomas. Oncogene. 4:1111–1116. 1989.PubMed/NCBI
|
|
7
|
De Herdt MJ and de Jong RJ Baatenburg: HGF
and c-MET as potential orchestrators of invasive growth in head and
neck squamous cell carcinoma. Front Biosci. 13:2516–2526. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kalyankrishna S and Grandis JR: Epidermal
growth factor receptor biology in head and neck cancer. J Clin
Oncol. 24:2666–2672. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rørth P: Collective cell migration. Annu
Rev Cell Dev Biol. 25:407–429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Neiva KG, Zhang Z, Miyazawa M, Warner KA,
Karl E and Nör JE: Cross talk initiated by endothelial cells
enhances migration and inhibits anoikis of squamous cell carcinoma
cells through STAT3/Akt/ERK signaling. Neoplasia. 11:583–593. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Friedl P and Wolf K: Plasticity of cell
migration: A multiscale tuning model. J Cell Biol. 188:11–19. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jorissen RN, Walker F, Pouliot N, Garrett
TP, Ward CW and Burgess AW: Epidermal growth factor receptor:
Mechanisms of activation and signalling. Exp Cell Res. 284:31–53.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Temam S, Kawaguchi H, El-Naggar AK,
Jelinek J, Tang H, Liu DD, Lang W, Issa JP, Lee JJ and Mao L:
Epidermal growth factor receptor copy number alterations correlate
with poor clinical outcome in patients with head and neck squamous
cancer. J Clin Oncol. 25:2164–2170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R,
Hammond EH, Fu KK and Milas L: Impact of epidermal growth factor
receptor expression on survival and pattern of relapse in patients
with advanced head and neck carcinoma. Cancer Res. 62:7350–7356.
2002.PubMed/NCBI
|
|
16
|
Bonner JA, Harari PM, Giralt J, Cohen RB,
Jones CU, Sur RK, Raben D, Baselga J, Spencer SA, Zhu J, et al:
Radiotherapy plus cetuximab for locoregionally advanced head and
neck cancer: 5-year survival data from a phase 3 randomised trial,
and relation between cetuximab-induced rash and survival. Lancet
Oncol. 11:21–28. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fung C and Grandis JR: Emerging drugs to
treat squamous cell carcinomas of the head and neck. Expert Opin
Emerg Drugs. 15:355–373. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ohnishi Y, Yasui H, Kakudo K and Nozaki M:
Cetuximab-resistant oral squamous cell carcinoma cells become
sensitive in anchorage-independent culture conditions through the
activation of the EGFR/AKT pathway. Int J Oncol. 47:2165–2172.
2015.PubMed/NCBI
|
|
19
|
Ohnishi Y, Yasui H, Kakudo K and Nozaki M:
Regulation of cell migration via EGFR signaling in oral squamous
cell carcinoma. Oncol Lett. 13:930–936. 2017.PubMed/NCBI
|
|
20
|
Birchmeier C, Birchmeier W, Gherardi E and
Woude GF Vande: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Benvenuti S and Comoglio PM: The MET
receptor tyrosine kinase in invasion and metastasis. J Cell
Physiol. 213:316–325. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma PC, Maulik G, Christensen J and Salgia
R: c-Met: Structure, functions and potential for therapeutic
inhibition. Cancer Metastasis Rev. 22:309–325. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peruzzi B and Bottaro DP: Targeting the
c-Met signaling pathway in cancer. Clin Cancer Res. 12:3657–3660.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hui AY, Meens JA, Schick C, Organ SL, Qiao
H, Tremblay EA, Schaeffer E, Uniyal S, Chan BM and Elliott BE: Src
and FAK mediate cell-matrix adhesion-dependent activation of Met
during transformation of breast epithelial cells. J Cell Biochem.
107:1168–1181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Henderson V, Smith B, Burton LJ, Randle D,
Morris M and Odero-Marah VA: Snail promotes cell migration through
PI3K/AKT-dependent Rac1 activation as well as PI3K/AKT-independent
pathways during prostate cancer progression. Cell Adhes Migr.
9:255–264. 2015. View Article : Google Scholar
|
|
26
|
Lin CW, Sun MS, Liao MY, Chung CH, Chi YH,
Chiou LT, Yu J, Lou KL and Wu HC: Podocalyxin-like 1 promotes
invadopodia formation and metastasis through activation of
Rac1/Cdc42/cortactin signaling in breast cancer cells.
Carcinogenesis. 35:2425–2435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rottner K and Stradal TE: Actin dynamics
and turnover in cell motility. Curr Opin Cell Biol. 23:569–578.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Usatyuk PV, Fu P, Mohan V, Epshtein Y,
Jacobson JR, Gomez-Cambronero J, Wary KK, Bindokas V, Dudek SM,
Salgia R, et al: Role of c-Met/phosphatidylinositol 3-kinase
(PI3k)/Akt signaling in hepatocyte growth factor (HGF)-mediated
lamellipodia formation, reactive oxygen species (ROS) generation,
and motility of lung endothelial cells. J Biol Chem.
289:13476–13491. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Michael M, Vehlow A, Navarro C and Krause
M: c-Abl, Lamellipodin, and Ena/VASP proteins cooperate in dorsal
ruffling of fibroblasts and axonal morphogenesis. Curr Biol.
20:783–791. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Law AL, Vehlow A, Kotini M, Dodgson L,
Soong D, Theveneau E, Bodo C, Taylor E, Navarro C, Perera U, et al:
Lamellipodin and the Scar/WAVE complex cooperate to promote cell
migration in vivo. J Cell Biol. 203:673–689. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takeshita A, Iwai S, Morita Y,
Niki-Yonekawa A, Hamada M and Yura Y: Wnt5b promotes the cell
motility essential for metastasis of oral squamous cell carcinoma
through active Cdc42 and RhoA. Int J Oncol. 44:59–68.
2014.PubMed/NCBI
|
|
32
|
Ma PC, Jagadeeswaran R, Jagadeesh S,
Tretiakova MS, Nallasura V, Fox EA, Hansen M, Schaefer E, Naoki K,
Lader A, et al: Functional expression and mutations of c-Met and
its therapeutic inhibition with SU11274 and small interfering RNA
in non-small cell lung cancer. Cancer Res. 65:1479–1488. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wehrle-Haller B and Imhof BA: Actin,
microtubules and focal adhesion dynamics during cell migration. Int
J Biochem Cell Biol. 35:39–50. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Krause M, Leslie JD, Stewart M, Lafuente
EM, Valderrama F, Jagannathan R, Strasser GA, Rubinson DA, Liu H,
Way M, et al: Lamellipodin, an Ena/VASP ligand, is implicated in
the regulation of lamellipodial dynamics. Dev Cell. 7:571–583.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hsu PC, You B, Yang YL, Zhang WQ, Wang YC,
Xu Z, Dai Y, Liu S, Yang CT, Li H, et al: YAP promotes erlotinib
resistance in human non-small cell lung cancer cells. Oncotarget.
7:51922–51933. 2016.PubMed/NCBI
|
|
36
|
Ritter CA and Arteaga CL: The epidermal
growth factor receptor-tyrosine kinase: A promising therapeutic
target in solid tumors. Semin Oncol. 30 Suppl 1:3–11. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kuhlmann CR, Schaefer CA, Fehsecke A, Most
AK, Tillmanns H and Erdogan A: A new signaling mechanism of
hepatocyte growth factor-induced endothelial proliferation. J
Thromb Haemost. 3:2089–2095. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wondergem R, Ecay TW, Mahieu F, Owsianik G
and Nilius B: HGF/SF and menthol increase human glioblastoma cell
calcium and migration. Biochem Biophys Res Commun. 372:210–215.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tan CS, Gilligan D and Pacey S: Treatment
approaches for EGFR-inhibitor-resistant patients with
non-small-cell lung cancer. Lancet Oncol. 16:e447–e459. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu X, Miyamoto S and Mekada E: Integrin α
2 β 1-dependent EGF receptor activation at cell-cell contact sites.
J Cell Sci. 113:2139–2147. 2000.PubMed/NCBI
|
|
41
|
Bill HM, Knudsen B, Moores SL, Muthuswamy
SK, Rao VR, Brugge JS and Miranti CK: Epidermal growth factor
receptor-dependent regulation of integrin-mediated signaling and
cell cycle entry in epithelial cells. Mol Cell Biol. 24:8586–8599.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Z: Transactivation of epidermal
growth factor receptor by G protein-coupled receptors: Recent
progress, challenges and future research. Int J Mol Sci.
17:E952016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Díaz B, Yuen A, Iizuka S, Higashiyama S
and Courtneidge SA: Notch increases the shedding of HB-EGF by
ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol.
201:279–292. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ohnishi Y, Inoue H, Furukawa M, Kakudo K
and Nozaki M: Heparin-binding epidermal growth factor-like growth
factor is a potent regulator of invasion activity in oral squamous
cell carcinoma. Oncol Rep. 27:954–958. 2012.PubMed/NCBI
|
|
45
|
Dulak AM, Gubish CT, Stabile LP, Henry C
and Siegfried JM: HGF-independent potentiation of EGFR action by
c-Met. Oncogene. 30:3625–3635. 2011. View Article : Google Scholar : PubMed/NCBI
|