Introduction
HCC is the most common primary liver tumor and one
of the most frequent malignant neoplasms worldwide (1). HCC is often not apparent, and thus
many patients are diagnosed at advanced stages, which means that
treatment is often less effective (2). These limited treatment options
highlight the need to clarify the mechanisms of HCC development and
identify early disease biomarkers and new therapeutic targets.
Chemokines play an essential role in tumor progression, and they
have been reported to sustain tumor cell growth, induce
angiogenesis and facilitate tumor escape through evasion of immune
surveillance (3–6). Several studies have confirmed that the
molecular pathogenesis of HCC involves chemokine/chemokine
receptors that participate in tumorigenesis and metastasis
(7–9). CXCR4 is the most common chemokine
receptor shown to be overexpressed in several cancers and to play
an important role in tumorigenesis (10–12).
Indeed, the level of CXCR4 expression serves as a prognostic
measure of disease progression and survival. Although the receptor
has been shown to be involved in HCC carcinogenesis, the specific
role of CXCR4 in HCC remains incompletely understood. Thus, the
molecular mechanism of CXCR4 in the progression of HCC warrants
further investigation.
The recently developed genome-editing technique
based on CRISPR/Cas9 is a broadly applicable approach for the
efficient modification of essentially any sequence of interest in
living cells or organisms (13).
This system has the capability to quickly and efficiently disrupt
genes, which often leads to translational termination and loss of
gene function. In the present study, we targeted CXCR4 by
CRISPR/Cas9 in the hepatoma cell line HepG2 to further elucidate
the function of CXCR4 in HepG2 cells both in vitro and in
vivo.
Materails and methods
Transfection and selection of stable
luciferase-expressing HepG2 cells
The hepatoma cell line HepG2 was cultured in
Dulbeccos modified Eagles medium (DMEM; Gibco, Carsbad, CA, USA)
supplemented with 10% fetal bovine serum (FBS) and incubated in a
37°C humidified atmosphere containing 5% CO2. The
pGL4.51(luc2/CMV/Neo) vector (Promega, Madison, WI, USA) was
transfected into the cells using Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA) according to the manufacturers instructions. At
24 h after transfection, the cell solution was diluted 1:10 and
regenerated. A total of 400 mg/ml G418 (Invitrogen) was added to
the 10% DMEM culture medium for selection after the cells adhered
to the plate. Limiting dilution was performed in a 96-well plate
for repeated colony selection.
CRISPR/Cas9-mediated downregulation of
CXCR4
The gRNA-coding cDNAs for targeting CXCR4 gene were
designed and synthesized to make the CXCR4-gRNA-Cas9 constructs.
The primers including 20 bp target sequence and BsmBI sticky
end were annealed and inserted into the pGK1.2 plasmid (Genloci
Biotechnologies) digested with BsmBI (NEB). Primer sequences
are as follows: sgRNA1, forward, 5′-CACCGCACTTCAGATAACTACACCG-3′
and reverse 5′-AAACCGGTGTAGTTATCTGAAGTGC-3′; sgRNA2, forward,
5′-CACCGAAGCATGACGGACAAGTAC-3′ and reverse,
5′-AAACGTACTTGTCCGTCATGCTTC-3′; sgRNA3, forward,
5′-CACCGGGCAATGGATTGGTCATCC-3′ and reverse,
5′-AAACGGATGACCAATCCATTGCCC-3′. Approximately 10 µl of
CXCR4-gRNA-Cas9 plasmids (10 µg) was mixed with 1×106
HepG2 cells in a cuvette containing 90 µl Opti-MEM. The
transfection was performed under 150 V with a NEPA21 electroporator
(Nepa Gene, Co., Ltd., Chiba, Japan). The transfected cells were
transferred to a well of a 6-well plate and cultured at 37°C with 3
µg/ml puromycin. After 3 days, the medium was replenished without
the addition of puromycin, and the cells were maintained at 37°C
for a few more days before they were harvested for DNA extraction
and single-cell culture to select CXCR4-silenced cell clones.
T7E1 assay
Genomic DNA was prepared from wild-type HepG2 cells
(control) and HepG2 CXCR4-downregulated cells (CXCR4−)
with the Genomic DNA Extraction Mini kit (Tiangen Biotech, Co.,
Ltd., Beijing, China). The genomic region that encompasses the
target site was amplified by PCR, and its products were denatured
and annealed to form heteroduplex DNA. The primer sequences to
amplify the genomic region were as follows: forward,
5′-ACTATGGGAAAAGATGGG-3′ and reverse, 5′-ACTGCTGTAGAGGTTGACTG-3′;
The annealed DNA was treated with 5 units of T7E1 (ViewSolid
Biotechnology, Beijing, China) at 37°C for 15 min; the DNA was then
run on an agarose gel to separate the DNA fragments and identify
the mutant clones. The target rate was calculated based on the
intensities of DNA bands, which were proportionally measured by
grey scale technique.
Cell proliferation assay
Cell proliferation assay was used to evaluate cell
proliferation according to the manufacturer's instructions. The
cells were seeded in 96-well plates at 1×104 cells/well
and cultured for 24, 48, 72, 96, 120 and 144 h. A total of 20 µl of
CellTiter one solution reagent (Promega) was added to 100 µl of
medium in each well, the plates were incubated for 4 h at 37°C, and
the absorbance values were read by a 96-well plate reader at the
absorbance of 490 nm.
Western blot analysis
Cells from each group were collected, and proteins
were extracted and separated by SDS-PAGE and then transferred onto
PVDF membranes. After blocking with 1% BSA for 1 h at room
temperature, the membranes were incubated with antibodies specific
for CXCR4 (1:200; Abcam, Cambridge, ΜΑ, USA), Bcl2 (1:200; Abcam),
vimentin (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA),
E-cadherin (1:200; Santa Cruz Biotechnology), N-cadherin (1:200;
Santa Cruz Biotechnology), MDR1 (1:200; Santa Cruz Biotechnology)
and β-actin (1:1,000; Tiangen Biotech) overnight at 4°C. After 3
washes in TBST, the membranes were incubated with alkaline
phosphatase-conjugated secondary antibody (1:500; Tiangen Biotech)
at room temperature for 1 h. Then, the membranes were washed 3
times with TBST and imaged with a gel imaging system (Bio-Rad
Laboratories, Hercules, CA, USA).
Clonogenicity assay
The clonogenicity of the cells was analyzed by
colony-formation assay. The HepG2 cells with/without CXCR4
downregulation were plated onto a 12-well plate pre-coated with 1%
gelatin at a density of 1,000 cells/well. The cells were incubated
at 37°C for 10 days and then the colonies were stained by crystal
violet. The experiment was performed in triplicate.
Scratch cell migration assay
The scratch assay was used to observe cell
migration. HepG2 cells with/without CXCR4 downregulation were
cultured to complete confluence in 6-well plates, and a scratch was
made across the cell monolayer in each well with a 10-µl pipette
tip. Then, the cell monolayer was washed 3 times with
phosphate-buffered saline (PBS) and incubated in serum-free DMEM at
37°C with 5% CO2 for 24 or 48 h. The width of the
scratches was measured and the percentages by which the width
decreased were compared at 0, 24 and 48 h. The experiment was
performed in triplicate.
Invasion assay
Cell invasion assay was performed using the
Transwell chamber (8-µm; Corning Life Sciences, Corning, NY, USA).
The interior of the inserts precoated with 10 mg/ml growth
factor-reduced Matrigel (BD Biosciences, Franklin Lakes, NJ, USA).
Cells (2×104) were added to the interior of the inserts
in 0.2 ml of serum-free growth medium. Growth medium (0.8 ml)
supplemented with 10% bovine calf serum (BCS) was added to the
lower chamber. After incubation for 48 h at 37°C, the cells
attached to the upper surface of the filter were removed with a
cotton swab, and migrated cells on the lower surface of the filter
were fixed and stained for 15 min with 0.25% crystal violet
(Sigma-Aldrich, St. Louis, MΟ, USA), 10% formaldehyde and 80%
methanol, and then the inserts were washed five times with
ddH2O and photographed. Migrated cells were determined
by counting cells in five microscopic fields per well, and the
extent of migration was expressed as an average number of cells per
microscopic field. Cells were imaged by phase contrast
microscopy.
Quantitative PCR
Total RNA was extracted using TRIzol reagent
(Ambion, Austin TX, USA) according to the manufacturers
recommendations and was quantified by UV spectroscopy. To prepare
the RNA for PCR analysis, 2 µg total RNA was converted to cDNA
using the FastQuant RT kit with gDNase (Tiangen Biotech).
Quantitative PCR was then conducted with a SYBR-Green Master Mix
(Takara Bio, Dalian, China). The PCR reaction proceeded as follows:
95°C for 30 sec, followed by 40 cycles of 95°C for 30 sec, 60°C for
30 sec and 72°C for 30 sec. The gene expressions were normalized to
GAPDH. The primer sequences for the genes were as follows: GAPDH,
forward, 5′-CCAAGGTCATCCATGACAAC-3′ and reverse,
5′-TGTCATACCAGGAAATGAGC-3′; Snail, forward,
5′-GAAAGGCCTTCAACTGCAAA-3′ and reverse, 5′-TGACATCTGAGTGGGTCTGG-3′;
Slug, forward, 5′-AGATGCATATTCGGACCCAC-3′ and reverse,
5′-CCTCATGTTTGTGCAGGAGA-3′.
Chemosensitivity assay
HepG2 cells with/without CXCR4 downregulation were
seeded onto a 96-well plate (1×104 cells/well) and
incubated at 37°C for 24 h. The cells received fresh medium that
contained 1 µg/ml cisplatin, and the cells were continuously
incubated for an additional 48 h; the cells were then removed from
the incubator and treated with 1 µl luciferase substrate, which was
added to the medium for 10–15 min. The viability of the cells was
examined using a fluorescence imaging system (IVIS®
Spectrum; Caliper Life Sciences, Inc., Waltham, MA, USA). The
experiment was performed in triplicate.
Xenograft assay
HepG2 cells with/without CXCR4 down-regulation at a
density of 5×106 were subcutaneously injected into 4- to
6-week-old nude mice (3 mice per group). These mice were bred, and
the sizes of the neoplasms from the implanted cells were examined
weekly using the IVIS Spectrum system (Caliper Life Sciences,
Waltham, MA, USA). Mice were anesthetized by inhalation of 2%
isoflurane and then received an intra-peritoneal injection of
D-luciferin; bioluminescence images were obtained 15 min after the
luciferin injection. The bioluminescence signal was analyzed with
software after the placement of a small region of interest. The
mean light intensity was then measured within this region of
interest. The research protocol on animals was approved by the
Ethics Committee of the First Affiliated Taihe Hospital of Hubei
University of Medicine.
Statistical analysis
The results are presented as the mean ± standard
deviation (SD). The statistical significance of the differences was
tested with the Students t-test in Microsoft Office Excel. In the
comparisons, values of P<0.05 were considered statistically
significant.
Results
T7E1 analysis and selection of CXCR4
mutant cell clones
In order to genetically disrupt the CXCR4 allele, we
designed 3 gRNAs to target Cas9 to the conserved sites of human
CXCR4 gene (Fig. 1A) and generated
CXCR4-gRNA-Cas9 plasmids to deliver gRNAs into HepG2 cells. The
target region of the CXCR4 gene was amplified by PCR and then
digested with the T7E1 enzyme. We found that the three gRNAs
induced CXCR4 mutations at a rate of 23.5, 25 and 29.5%,
respectively (Fig. 1B). We chose #3
gRNA for the following experiment, different mutations were found
according to the gene sequencing of 30 single-cell clones (Fig. 1C); in particular, one cell clone
with triallelic CXCR4 mutations (CXCR4−) was selected by
gene sequencing (Fig. 1D). At least
three CXCR4 alleles were mutated in this HepG2 cell clone, and this
clone was therefore used for subsequent experiments.
Downregulation of CXCR4 inhibits the
proliferation of HepG2 cells
The impact of downregulating CXCR4 expression on
cell proliferation was detected by MTT. The number of proliferating
cells was determined by measuring the optical density (OD) value at
490 nm. The detection results indicated that the speed of cell
proliferation was slower in the CXCR4− group than the
control group after cultured for 4 days (P<0.05; Fig. 2A). We also detected the
proliferation and apoptosis associated protein Bcl2 by western blot
analysis, the result showed that downregulation of CXCR4 could
decrease the expression level of Bcl2 protein (Fig. 2B).
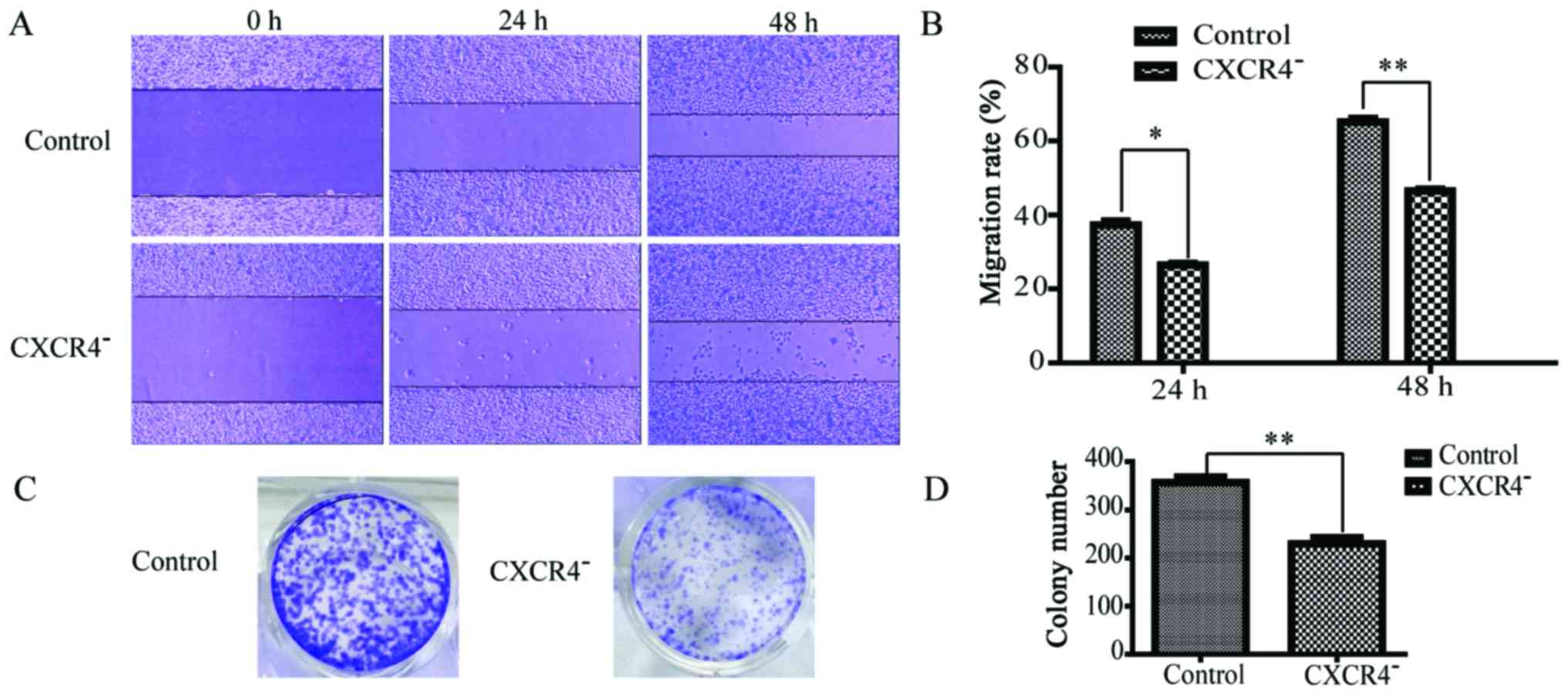
Downregulation of CXCR4 restricts the
migration and cloning efficiency of HepG2 cells
According to the scratch assay, the migration rates
of CXCR4− HepG2 cells at 24 and 48 h were 24±3 and
45±4%, respectively, while those for wild-type HepG2 cells were
36±4 and 64±7%, respectively (Fig. 3A
and B). This significant difference (P<0.05) indicated that
the migration of HepG2 cells was strongly inhibited by CXCR4
downregulation. In the cell colony formation assay, as shown in
Fig. 3C and D, the number of
colonies formed by CXCR4− HepG2 cells was 229±10, while
that formed by wild-type HepG2 cells was 356±14. This significant
difference (P<0.05) indicated that the clonogenicity of HepG2
cells was strongly inhibited by CXCR4 downregulation.
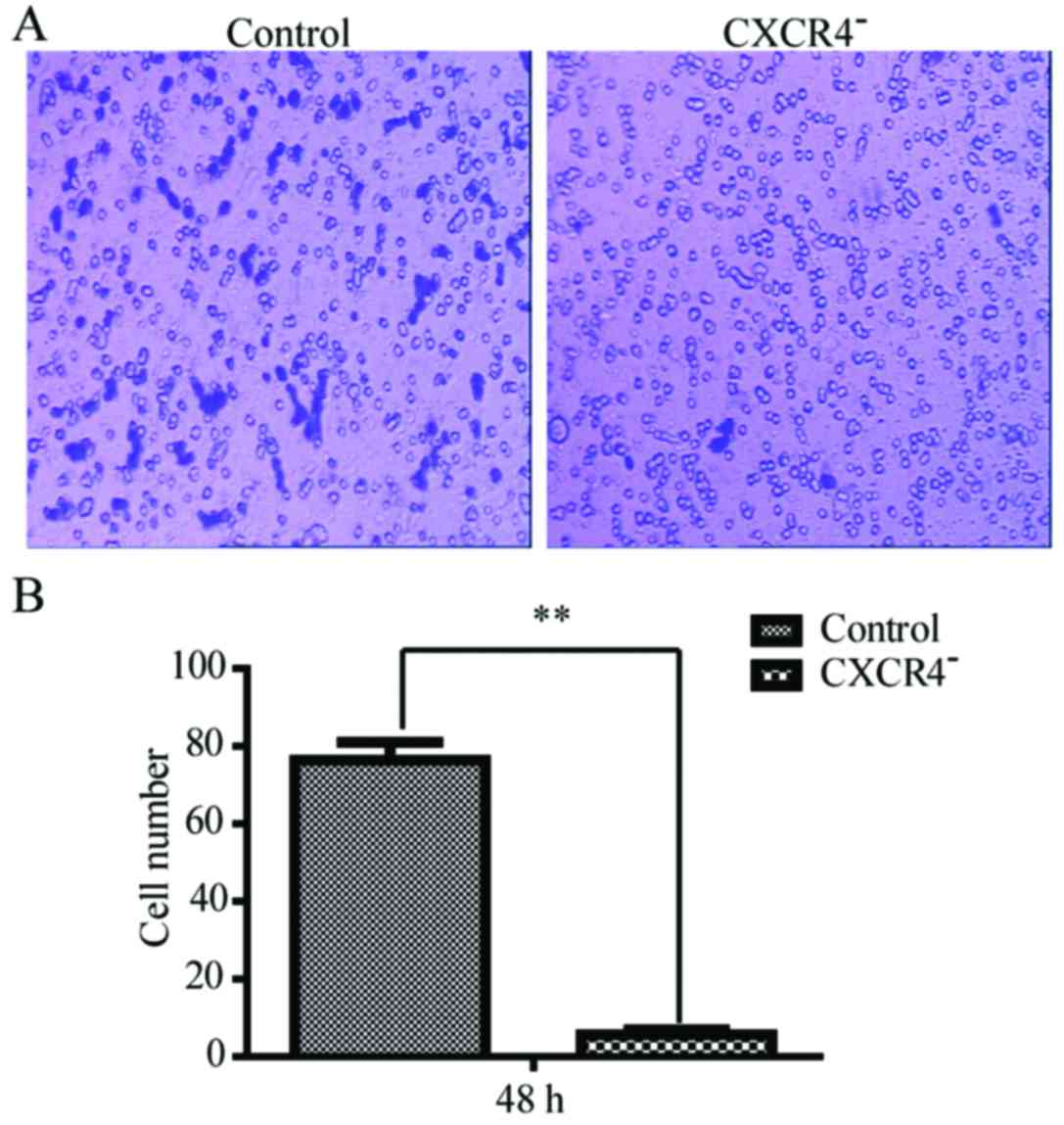
Downregulation of CXCR4 alleviates the
invasiveness of the HepG2 cells
Transwell cell migration assay was carried out to
investigate how the invasiveness of HepG2 cells was affected by
CXCR4 disruption. As shown in Fig.
4, the numbers of the cells that passed through the Matrigel at
48 h was 12±3 for CXCR4-HepG2 cells and 78±7 for the HepG2 cells.
The number for the downregulation of CXCR4 in HepG2 cells was
remarkably less than the wild-type (P<0.05).
Downregulation of CXCR4 in HepG2 cells
reverses the status of EMT
EMT is a critical event that can be observed during
tumor progression. Quantitative RT-PCR analysis revealed that the
expression levels of some EMT-regulated transcription factor genes,
such as Slug and Snail, were downregulated in CXCR4−
HepG2 cells (Fig. 5A and B). At the
protein level, upregulation of the epithelial marker E-cadherin and
downregulation of the mesenchymal markers vimentin and N-cadherin
were also observed (Fig. 5C).
Together, these changes suggested a reversal of EMT.
Downregulation of CXCR4 increases the
sensitivity of HepG2 cells to cisplatin in vitro
The chemosensitivity of the HepG2 and the
CXCR4− HepG2 cells was compared according to their
viability after exposure to cisplatin. As shown in Fig. 6A and B, CXCR4− HepG2
cells were more sensitive to cisplatin. MDR1, which is an important
indicator of drug resistance in chemotherapy, was also
significantly downregulated in CXCR4− HepG2 cells
(Fig. 6C).
Downregulation of CXCR4 decreases the
growth of HepG2 xenograft tumor in nude mice
To further investigate the inhibitory effect of
CXCR4− HepG2 cells on tumor cell growth in vivo,
we used a xenograft cancer model. Either CXCR4− HepG2
cells or wild-type HepG2 cells were injected subcutaneously into
nude mice, and the sizes of the neoplasms that formed from the
implanted HepG2 cells were measured weekly (Fig. 7A). Based on our results, the sizes
of the neoplasms formed from the CXCR4− HepG2 cells were
significantly smaller than those formed from wild-type cells
(Fig. 7B).
Discussion
The development of a malignant tumor is a complex
process that involves multiple factors and stages as well as
malfunctions or mutations in a variety of genes. CRISPR/Cas9 is a
useful tool that can be used to study the function of genes
(14). Although CXCR4 has been
shown to be involved in many malignant human tumors, few studies
have investigated the effects of the directed inhibition of CXCR4
in HCC. To gain additional insight into the effects of CXCR4 in
HepG2 cells, we selectively knocked down CXCR4 expression using
CRISPR/Cas9 and observed the effects both in vitro and in
vivo. In the present study, CXCR4 was effectively disrupted by
CRISPR/Cas9, and the expression of CXCR4 was remarkably
downregulated and the cell proliferation was inhibited.
The grade of malignancy is closely related to the
migration clonogenicity and invasion of tumor cells. In the present
study, scratch assays showed that the migration and invasiveness of
CXCR4− HepG2 cells were significantly restricted, and
colony formation assays showed that the clonogenicity of HepG2
cells with knocked down CXCR4 expression was decreased. These
findings suggested that CXCR4− HepG2 cells were less
malignant than their wild-type counterparts.
EMT may indicate a progression toward malignancy,
with increased potential for invasion and metastasis (15–17).
During this process, cancer cells acquire a fibroblast-like
phenotype (18). However, in the
present study, this progression seemed to be markedly reversed by
the downregulation of CXCR4. After downregulation of CXCR4 in HepG2
cells, the expression levels of several genes that are critical for
progression from an epithelial to a mesenchymal phenotype were
altered. Specifically, we observed the downregulation of
mesenchymal markers such as vimentin and snail and the upregulation
of epithelial markers such as E-cadherin. These data suggested that
CXCR4 may play a critical role in the process of EMT. An
understanding of the molecular mechanisms responsible for EMT and
the discovery of approaches to reverse this process should be very
important for the development of new therapeutic strategies against
HCC.
The cytotoxicity of cisplatin to normal tissues and
the acquired resistance of cancer cells to this drug have reduced
its clinical efficacy (19).
However, the exact mechanism of cisplatin resistance is not clear.
MDR1 is the most important indicator for chemoresistance (20,21).
Our experiments result revealed that tumor CXCR4 can modulate
cisplatin sensitivity by influence the expression level of MDR1 and
the downregulation of CXCR4 in HepG2 cells increased their
sensitivity to cisplatin in vitro.
In conclusion, we used CRISPR/Cas9 to efficiently
dowregulate CXCR4 expression, which led to a marked decrease in the
grade of malignancy of HCC in vitro and in vivo.
CRISPR/Cas9-mediated genome engineering may be a good way to study
the function of the potential oncogenes on the genome level and
provide a rapid avenue for functional cancer genomics study in the
future.
Acknowledgements
This study was supported by the National Natural
Science Foundation of Hubei province (2012FFA037), the Natural
Science Foundation of Hubei Provincial Department of Education
(Q20162110), the Key Science and Technology Project of Hubei
Province (2016ACA157) and the National Natural Science Foundation
of China (81641028).
References
|
1
|
Flores A and Marrero JA: Emerging trends
in hepatocellular carcinoma: focus on diagnosis and therapeutics.
Clin Med Insights Oncol. 8:71–76. 2014.PubMed/NCBI
|
|
2
|
Shinoda M, Kishida N, Itano O, Ei S, Ueno
A, Kitago M, Abe Y, Hibi T, Yagi H, Masugi Y, et al: Long-term
complete response of advanced hepatocellular carcinoma treated with
multidisciplinary therapy including reduced dose of sorafenib: Case
report and review of the literature. World J Surg Oncol.
13:1442015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gassmann P, Haier J, Schlüter K,
Domikowsky B, Wendel C, Wiesner U, Kubitza R, Engers R, Schneider
SW, Homey B, et al: CXCR4 regulates the early extravasation of
metastatic tumor cells in vivo. Neoplasia. 11:651–661. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hattermann K, Holzenburg E, Hans F, Lucius
R, Held-Feindt J and Mentlein R: Effects of the chemokine CXCL12
and combined internalization of its receptors CXCR4 and CXCR7 in
human MCF-7 breast cancer cells. Cell Tissue Res. 357:253–266.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH,
Wang Z, Huang XW, Fan J and Zhou J: Overexpression of CXCL5
mediates neutrophil infiltration and indicates poor prognosis for
hepatocellular carcinoma. Hepatology. 56:2242–2254. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wendt MK, Johanesen PA, Kang-Decker N,
Binion DG, Shah V and Dwinell MB: Silencing of epithelial CXCL12
expression by DNA hypermethylation promotes colonic carcinoma
metastasis. Oncogene. 25:4986–4997. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schimanski CC, Bahre R, Gockel I, Müller
A, Frerichs K, Hörner V, Teufel A, Simiantonaki N, Biesterfeld S,
Wehler T, et al: Dissemination of hepatocellular carcinoma is
mediated via chemokine receptor CXCR4. Br J Cancer. 95:210–217.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen RX, Song HY, Dong YY, Hu C, Zheng QD,
Xue TC, Liu XH, Zhang Y, Chen J, Ren ZG, et al: Dynamic expression
patterns of differential proteins during early invasion of
hepatocellular carcinoma. PLoS One. 9:e885432014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiang ZL, Zeng ZC, Tang ZY, Fan J, Zhuang
PY, Liang Y, Tan YS and He J: Chemokine receptor CXCR4 expression
in hepatocellular carcinoma patients increases the risk of bone
metastases and poor survival. BMC Cancer. 9:1762009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ehtesham M, Winston JA, Kabos P and
Thompson RC: CXCR4 expression mediates glioma cell invasiveness.
Oncogene. 25:2801–2806. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Andre F, Xia W, Conforti R, Wei Y, Boulet
T, Tomasic G, Spielmann M, Zoubir M, Berrada N, Arriagada R, et al:
CXCR4 expression in early breast cancer and risk of distant
recurrence. Oncologist. 14:1182–1188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Blot E, Laberge-Le Couteulx S, Jamali H,
Cornic M, Guillemet C, Duval C, Hellot MF, Pille JY, Picquenot JM
and Veyret C: CXCR4 membrane expression in node-negative breast
cancer. Breast J. 14:268–274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hsu PD, Lander ES and Zhang F: Development
and applications of CRISPR-Cas9 for genome engineering. Cell.
157:1262–1278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gori JL, Hsu PD, Maeder ML, Shen S,
Welstead GG and Bumcrot D: Delivery and specificity of CRISPR-Cas9
genome editing technologies for human gene therapy. Hum Gene Ther.
26:443–451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Katoh M: Epithelial-mesenchymal transition
in gastric cancer (Review). Int J Oncol. 27:1677–1683.
2005.PubMed/NCBI
|
|
16
|
Rhim AD, Mirek ET, Aiello NM, Maitra A,
Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK,
Vonderheide RH, et al: EMT and dissemination precede pancreatic
tumor formation. Cell. 148:349–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Creighton CJ, Chang JC and Rosen JM:
Epithelial-mesenchymal transition (EMT) in tumor-initiating cells
and its clinical implications in breast cancer. J Mammary Gland
Biol Neoplasia. 15:253–260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakayama K, Kanzaki A, Ogawa K, Miyazaki
K, Neamati N and Takebayashi Y: Copper-transporting P-type
adenosine triphosphatase (ATP7B) as a cisplatin based
chemoresistance marker in ovarian carcinoma: Comparative analysis
with expression of MDR1, MRP1, MRP2, LRP and BCRP. Int J Cancer.
101:488–495. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Su F, Ouyang N, Zhu P, Ouyang N, Jia W,
Gong C, Ma X, Xu H and Song E: Psychological stress induces
chemoresistance in breast cancer by upregulating mdr1. Biochem
Biophys Res Commun. 329:888–897. 2005. View Article : Google Scholar : PubMed/NCBI
|