Introduction
Ultraviolet (UV) radiation exposure is the primary
risk factor underlying premature skin aging (photoaging) and skin
carcinogenesis (photocarcinogenesis), with the development of
cutaneous melanoma, squamous cell carcinoma (SCC), and basal cell
carcinoma (BCC) all being dependent upon UV radiation exposure
(1,2). Unfortunately, preventive topical
therapies (e.g., sunscreens and topical antioxidants) are not
sufficiently effective in shielding skin cells from the
DNA-damaging effects of UV radiation (3,4).
Among the various DNA lesions created by UV
radiation exposure, DNA double-strand breaks (DSBs) are
particularly potent, since they can produce substantial losses in
DNA integrity and are, therefore, more cytotoxic than other DNA
lesions (5). Moreover, substandard
DSB repair can result in DNA mutations or chromosomal
rearrangements, which can both result in de novo
carcinogenesis (5). For this
reason, mounting research has focused on the role of DNA damage
response pathways in DSB repair in order to develop novel
chemotherapeutic targets for photoaging and photocarcinogenesis
(3,6).
One DNA damage response protein in particular,
tumor-suppressor p53-binding protein 1 (53BP1), has been shown to
play a key role in DSB repair by regulating the cellular mechanism
by which DSBs are repaired (7).
Specifically, 53BP1 plays a central role in the choice between 2
DSB repair pathways: homologous recombination (HR) or
non-homologous end joining (NHEJ) (7). As 53BP1 inhibits an early
rate-limiting step in the HR pathway and blocks recruitment of the
HR pathway-promoting BRCA1 at DSBs, 53BP1 acts to promote NHEJ as
the DSB repair pathway of choice (7,8). As
NHEJ DSB repair is the more constitutively active and efficient
pathway for DSB repair, inappropriate regulation of 53BP1-mediated
NHEJ can lead to genomic instability and carcinogenesis (9).
On this basis, nuclear localization of 53BP1 at UV
radiation-induced foci is necessary for effective NHEJ DSB repair
(10). Mechanistically, 53BP1
expression is promoted by the intermediate filament protein lamin
A, a gene product of the LMNA gene (7). Accordingly, lamin A deficiency (while
preserving overall DNA damage response activation) results in NHEJ
DSB repair inhibition from decreased nuclear 53BP1 accumulation at
UV radiation-induced foci as a product of heightened 53BP1
degradation (10), while
artificially reconstituting 53BP1 in the presence of lamin A
deficiency has been shown to rescue NHEJ DSB repair activity
(10). This evidence suggests that
lamin A deficiency effectively inhibits NHEJ DSB repair activity
through the decrease of the expression of 53BP1.
Progerin, an aberrant UVA radiation-induced splicing
variant of the LMNA gene transcript (11), has been shown to be a potent lamin A
binding partner that decreases nucleoplasmic lamin A expression
(12). Notably, a recent study by
Waldman et al revealed that progerin overexpression
negatively impacted NHEJ DSB repair as opposed to HR DSB repair
(13). On the basis of the
aforementioned evidence, in the present study we hypothesized that
aberrant progerin upregulation in response to UVA radiation may
adversely affect 53BP1-mediated NHEJ DSB repair in human
keratinocytes.
Materials and methods
Ethics statement
Approval for the present study was obtained from the
Ethics Committee of the First Affiliated Hospital of Chongqing
Medical University (Chongqing, China). Prior informed written
consent was obtained from adult participants, and from the parents
or legal guardians of the human neonatal subjects. Standards from
the Guide for the Care and Use of Laboratory Animals [8th edition,
National Institutes of Health (NIH), Bethesda, MD, USA] were
followed for the present study.
Adult skin sampling
Study participants were recruited from outpatient
clinics affiliated with the First Affiliated Hospital of Chongqing
Medical University. Specifically, clinical physicians identified
adult patients (aged 18–79 years) with a histologically confirmed
BCC tumor diagnosed within a previous 6-month period for
recruitment. Candidates were summarily excluded when they were
immunocompromised, cognitively impaired, or unable to provide
informed consent. BCC tumors and matching normal skin tissue
samples from the recruited participants (n=200) were surgically
excised and stored at −80°C until subsequent analysis.
Primary culture of neonatal
keratinocytes
Newborn foreskin samples (n=9) were used as a source
of primary human neonatal keratinocytes as previously described
(14). Trypsinization was performed
on the proliferating primary cultures, and EpiLife culture medium
containing human keratinocyte growth supplement (Life Technologies,
Gaithersburg, MD, USA) was used to amplify the keratinocytes into
secondary cultures as previously described (15).
Monolayer cultures were produced using passage 3
(P3) keratinocyte cultures as previously described (15). Briefly, when cells attained a 60%
density, trypsinization was used to harvest the keratinocytes, and
the cells were subsequently plated at 5,000 cells/cm2.
Plated cultures were incubated to develop a keratinocyte monolayer.
At 50% confluence, the medium was replaced with an
EpiLife®-based medium free of growth factors and
hormones [EpiLife® medium with only amino acids,
antibiotics and hydrocortisone (Invitrogen-Cascade Biologics,
Mansfield, UK)] according to the autocrine conditions.
Keratinocytes were proliferated until confluence, at which point
expression of the keratinocyte differentiation markers involucrin
and keratin 10 were assssed.
UVA irradiation
UVA irradiation was performed as previously
described (11). Irradiations were
performed on culture plates resting in a temperature-regulated
water bath. Growth-arrested cells were repeatedly irradiated once
daily over a period of 7 days. A metal halogenide UVA lamp with an
infrared radiation-specific filter (2 kW; SELLAS Sunlight,
Ennepetal, Germany) was used for UVA irradiation at 5 and 10
J/cm2. The emission spectrum of the lamp ranged from
wavelengths of 335–440 nm with a peak at 375 nm.
Lentiviral transduction
The silencing of progerin was achieved via
transduction of lentiviral particles [Mission TRC short hairpin RNA
(shRNA) lentiviral transduction particles (Sigma-Aldrich, St.
Louis, MO, USA)] as previously described with minor modifications
(15). These particles contained
genes expressing resistance to puromycin and an shRNA under U6
promoter control. Two shRNAs were used: a previously described
anti-progerin shRNA (16), and a
non-mammalian shRNA control with no known target. Lentiviruses were
used to transduce keratinocytes at a 10-fold multiplicity of
infection (MOI). This was accomplished in the presence of 4 µg/ml
of protamine sulfate (Sigma-Aldrich). At 24 h, the culture medium
was exchanged for fresh medium containing 2 µg/ml of puromycin
(Sigma-Aldrich) for purposes of selection. When a 60% density was
achieved, harvesting by trypsinization was performed, and cells
were seeded in 6-well dishes for proliferation. Cells were finally
evaluated for appropriate mRNA expression.
53BP1 overexpression was performed as previously
described with minor modifications (17). A plasmid containing human 53BP1 cDNA
was purchased from Addgene (Cambridge, MA, USA). The 53BP1 cDNA
sequence was excised from the parental plasmid, and then ligated
into the lentiviral pCDH1-MCS1-EF1-copGFP expression vector (System
Biosciences, Mountain View, CA, USA). An empty null vector (NV) was
used as a negative control. Pseudoviral particles were amplified in
293TN cells with the pPACKH1-GAG packaging plasmid, pPACKH1-REV
(System Biosciences), and the pVSV-G envelope plasmid. Then, the
viral particles were concentrated, and resuspended in EpiLife-based
medium-free of growth factors and hormones for transduction into
the cultured neonatal keratinocytes.
Quantitative reverse transcription
PCR
Quantitative reverse transcription PCR (qRT-PCR) was
performed as previously described with minor modifications
(15). A Total RNA Isolation kit
(Tiangen, Beijing, China) was used to extract total RNA, and the
concentration was quantified with a NanoDrop 1000 UV/Vis
spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
SuperScript III RNase H reverse transcriptase (Invitrogen,
Carlsbad, CA, USA) was applied to reverse-transcribe RNA into cDNA.
The resulting cDNA was prepped with FastStart Universal SYBR-Green
Master (Roche, Basel, Switzerland) as well as sense and antisense
primers (300 nM; Sigma-Aldrich), which then underwent amplification
on an ABI 7300 Real-Time PCR System (Applied Biosystems, Foster
City, CA, USA). The following primers were applied: lamin A
forward, 5′-TCTTCTGCCTCCAGTGTCACG-3′ and reverse,
5′-AGTTCTGGGGGCTCTGGGT-3′; progerin forward,
5′-ACTGCAGCAGCTCGGGG-3′ and reverse 5′-GGCTCTGGGCTCCTGAGCC-3′;
53BP1 forward, 5′-GCCTGATCAATGGACCCTACTGGAAGTCAGG-3′, and reverse,
5′-CCGCTCGAGTTAGTGAGAAACATAATCGTGTTT-3′; and β-actin forward,
5′-GGAGACAAGCTTGCTCATCACCATTGGCAATGAGCG-3′, and reverse,
5′-GCGAATTCGAGCTCTAGAAGCATTTGCGGTGGACG-3′. β-actin expression
levels were used in the normalization of the target mRNAs.
Western blotting
Western blotting was performed as previously
described with minor modifications (15). Lysis buffer (consisting of 62.5 mM
Tris-HCl, 8.7% glycerol, 2% SDS and 0.2% dithiothreitol;
Sigma-Aldrich) was used to extract proteins from keratinocytes. The
Pierce 660 nm Protein Assay Reagent (cat. no. 22660) in conjunction
with anti-SDS (Thermo Scientific) was used to assess protein
concentrations. The separation of proteins (20 µg) was performed
with 10% SDS-PAGE and then the proteins were transferred to
polyvinylidene fluoride (PVDF) membranes. A solution of 5% powdered
milk diluted in PBS and 0.1% Tween-20 was used to saturate the
membranes. The membranes were then incubated for 1 h with the
following primary antibodies (all diluted 1:1,000): anti-lamin A,
anti-lamin A/C (both from Santa Cruz Biotechnology, Santa Cruz, CA,
USA), anti-progerin, anti-53BP1 and anti-β-actin (all from Abcam,
Cambridge, MA, USA). The washed membranes were incubated for 1 h
with the appropriate HRP-conjugated secondary antibodies (diluted
1:5,000; Boster Company, Wuhan, China). The ECL Western Blotting
Detection kit (Applygen, Beijing, China) was used for
chemiluminescent signal detection.
Progerin-lamin A interaction
assay
Direct interaction of progerin and lamin A in
cultured neonatal keratinocytes was evaluated as previously
described (12). Briefly,
streptavidin-bead precleared cell lysates were sequentially
incubated with biotin-JH4 or biotin and streptavidin beads. Then,
western blotting was performed on the precipitated materials. To
block the interaction of progerin and lamin A, a 24-h treatment
with JH4 (5 µM) was performed. The farnesyltransferase inhibitor
FTI-277 [which does not affect progerin-lamin A binding (12)] was applied as a negative control for
JH4. Cell lysates were subjected to immunoprecipitation (IP) with a
lamin A-specific antibody (Santa Cruz Biotechnology) followed by
immunoblotting with a progerin-specific antibody (Abcam). A cell
line of untreated neonatal keratinocytes was applied as a negative
control for progerin expression.
Neutral comet assays
The CometSlide assay kit (Trevigen, Gaithersburg,
MD, USA) was used in neutral comet assay procedures as previously
described with minor modifications (10). Irradiation doses were set at 10
J/cm2 at an incubation temperature of 37°C. Incubation
times were varied (0, 30, 60, 90, 120 and 150 min) to allow for the
repair of DNA damage. Agarose was used to embed the keratinocytes,
which were then lysed and treated with neutral electrophoresis.
Previous to image analysis, the keratinocytes underwent tethidium
bromide staining and were examined using fluorescence microscopy.
The result of single-cell electrophoresis was a comet-shaped DNA
distribution; an intact and high molecular weight DNA constitute
the comet head, while the tail consists of leading ends of
migrating fragments. The olive comet moment (OCM) was determined by
multiplying the percentage of tail DNA by the displacement between
the head and tail distribution means (10). The OCM was calculated via
CometScore™ version 1.5 (TriTek Corp., Sumerduck, VA, USA). A total
of 25–30 comets/sample were analyzed in each experiment.
Results
In the present study, we first investigated whether
human keratinocytes display dysregulated progerin expression as a
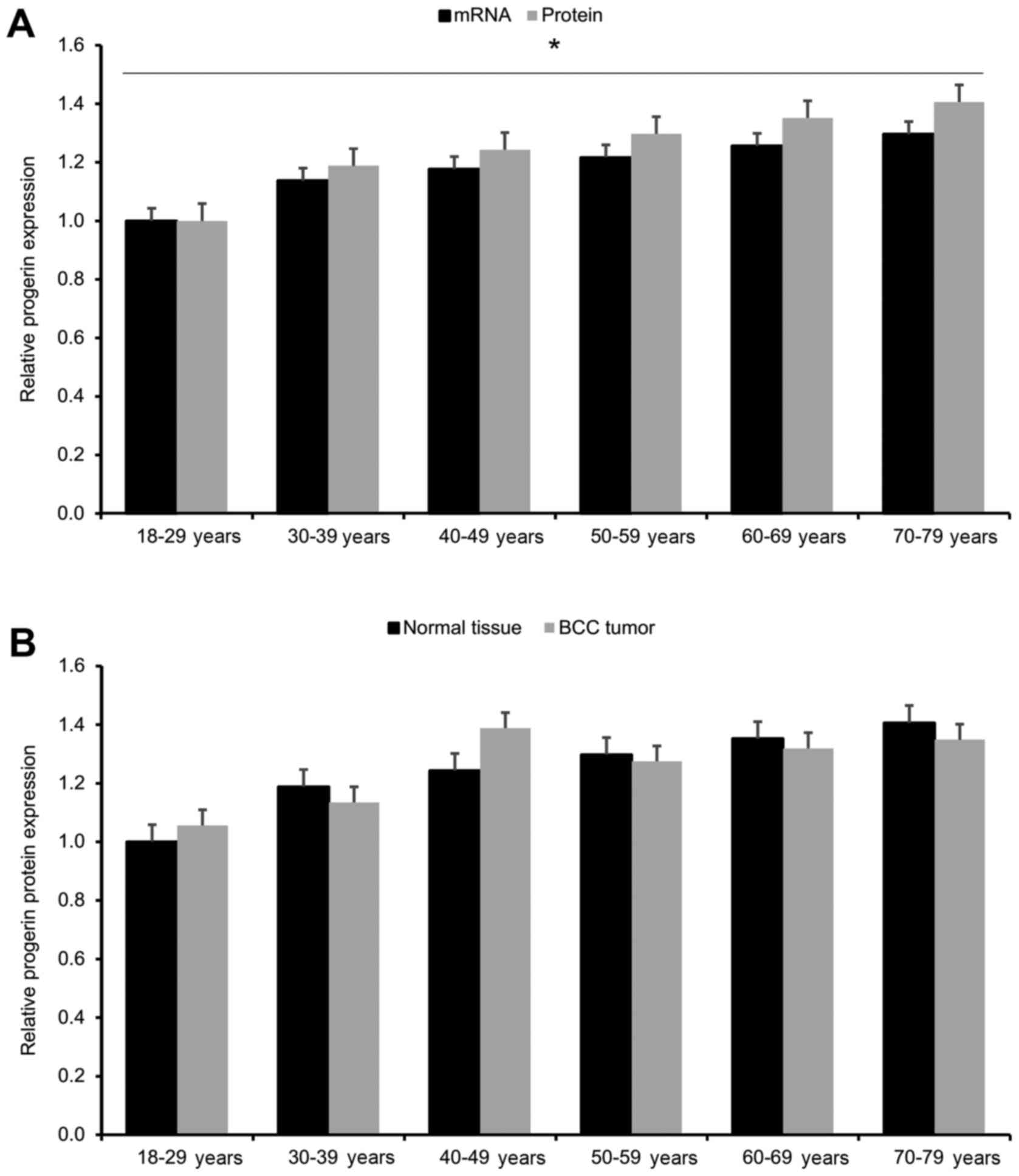
function of advancing age and BCC status. We found that progerin
expression in human keratinocytes gradually increased with
advancing age (p<0.05; Fig. 1A).
However, upon comparing each age group, we found no significant
differences in progerin protein expression between BCC tumors and
matched normal tissues across all age groups (p<0.05; Fig. 1B). These findings indicate that
progerin upregulation in human keratinocytes is associated with
advancing age, not BCC status.
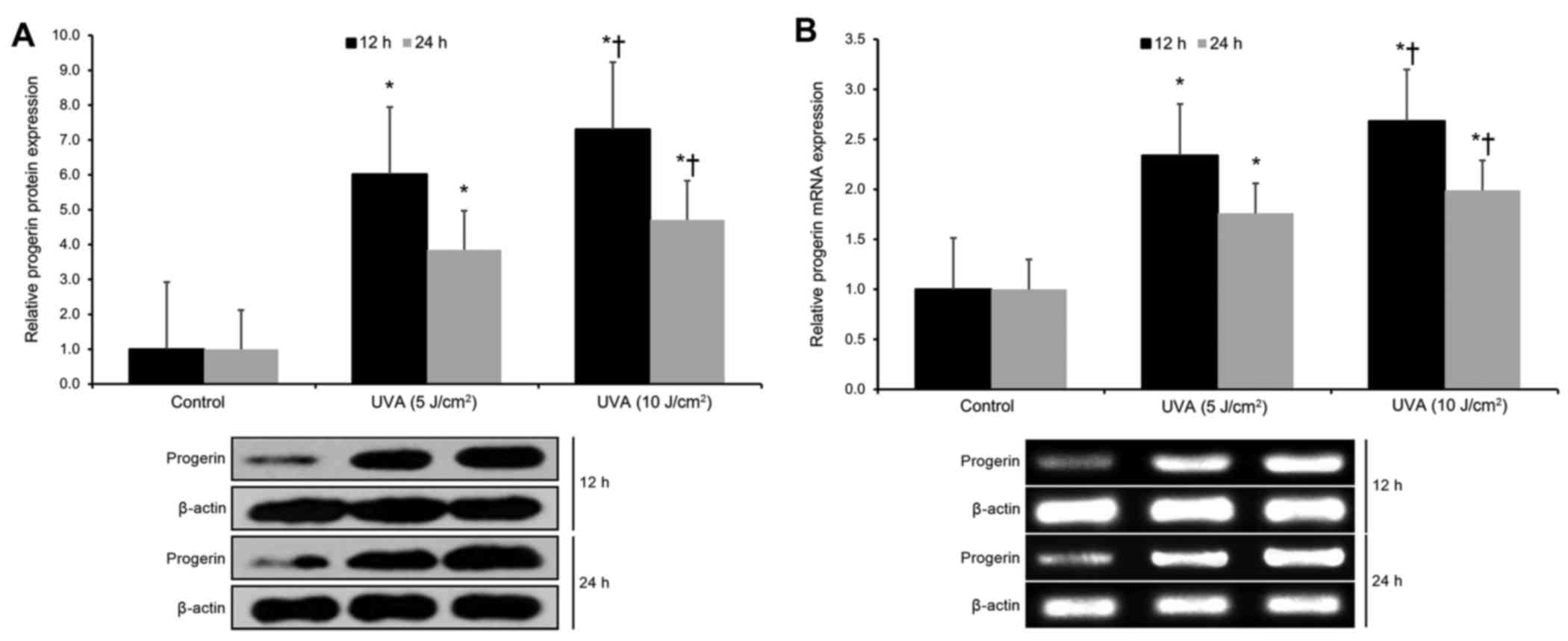
Next, we investigated the effects of UVA radiation
on progerin expression in cultured human neonatal keratinocytes. We
found that UVA exposure (at both 5 and 10 J/cm2 doses)
resulted in significant upregulation of progerin protein expression
at 12 and 24 h post-irradiation (p<0.05; Fig. 2A). Moreover, UVA exposure (at both 5
and 10 J/cm2 doses) resulted in significant upregulation
of progerin mRNA expression at 12 and 24 h post-irradiation
(p<0.05; Fig. 2B). These
findings indicate that UVA exposure significantly upregulates
progerin expression in human keratinocytes by favoring alternative
LMNA gene transcript splicing.
Next, we investigated the effects of UVA radiation
on lamin A expression in cultured human neonatal keratinocytes. We
found that UVA exposure (at both 5 and 10 J/cm2 doses)
resulted in significant downregulation of free (unbound) lamin A
protein expression at 12 and 24 h post-irradiation (p<0.05;
Fig. 2C). However, UVA exposure (at
both 5 and 10 J/cm2 doses) did not significantly affect
lamin A mRNA expression at 12 and 24 h post-irradiation (p>0.05;
Fig. 2D). These findings indicate
that UVA exposure significantly downregulates lamin A expression in
human keratinocytes by decreasing free lamin A protein levels (as
opposed to downregulating lamin A transcription).
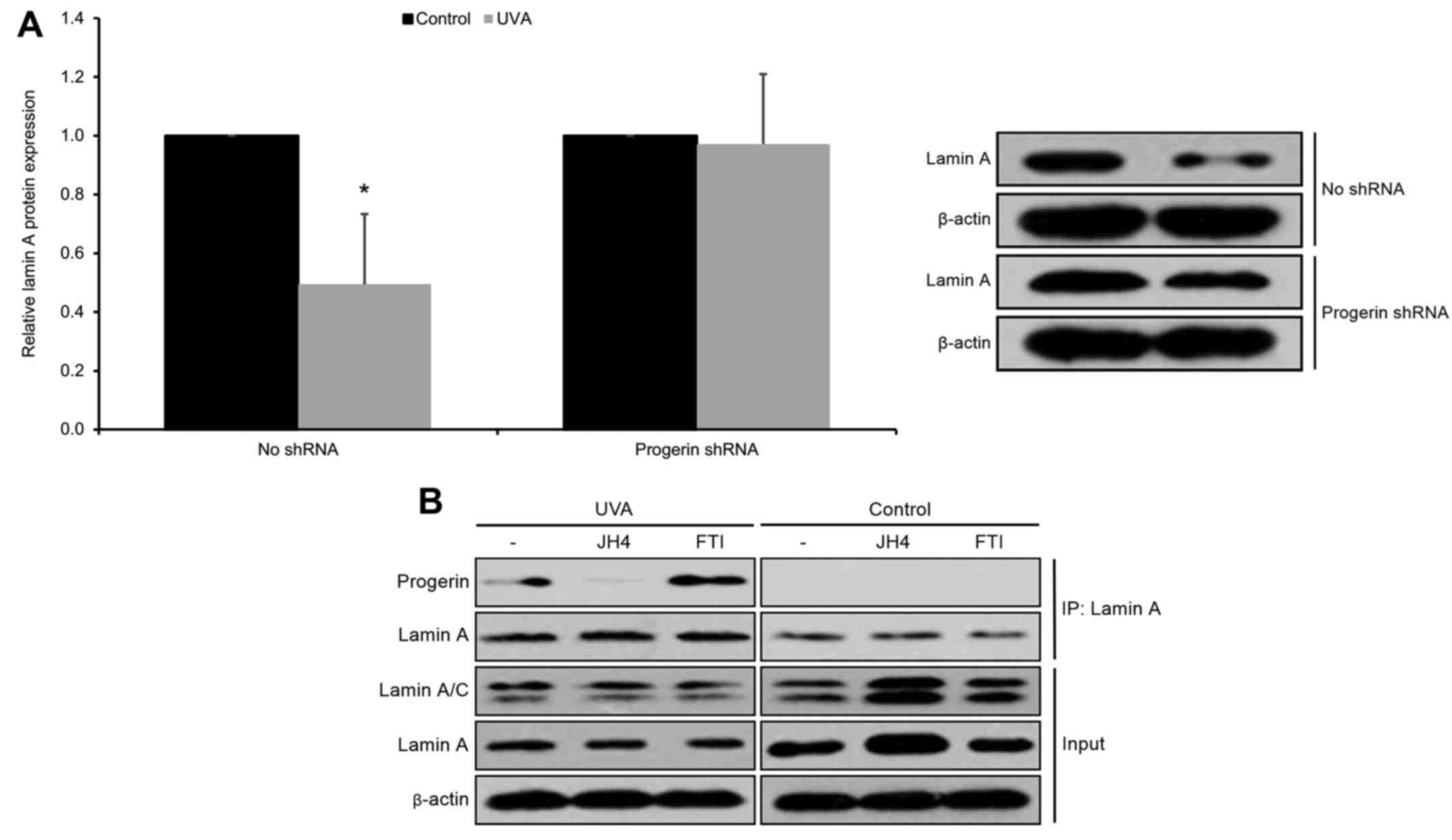
In order to further investigate the mechanism by
which UVA exposure decreases free lamin A protein levels in human
keratinocytes, we investigated the effects of silencing progerin on
free lamin A protein expression in UVA-irradiated cultured human
neonatal keratinocytes. We found that silencing of progerin (via a
progerin-specific shRNA) was able to rescue UVA-induced free lamin
A protein downregulation (p<0.05; Fig. 3A). From these data, we hypothesized
that progerin-lamin A interaction may result in the observed
downregulation in free lamin A protein levels in UVA-irradiated
keratinocytes. In order to test this hypothesis, we selected the
chemical compound JH4, which has been previously shown to prevent
progerin-lamin A binding in HGPS cells (12). We then ascertained that JH4 blocked
the interaction of progerin and lamin A in cultured human
keratinocytes (Fig. 3B). Next, we
demonstrated that blocking progerin-lamin A interaction by JH4 was
able to rescue UVA-induced lamin A protein downregulation
(p<0.05; Fig. 3C). As a control
experiment, we ascertained that JH4 administration produced no
significant effects on lamin A mRNA levels (p>0.05; Fig. 3D). These findings indicate that UVA
exposure significantly decreases free lamin A protein levels in
human keratinocytes through the promotion of progerin-lamin A
complex formation.
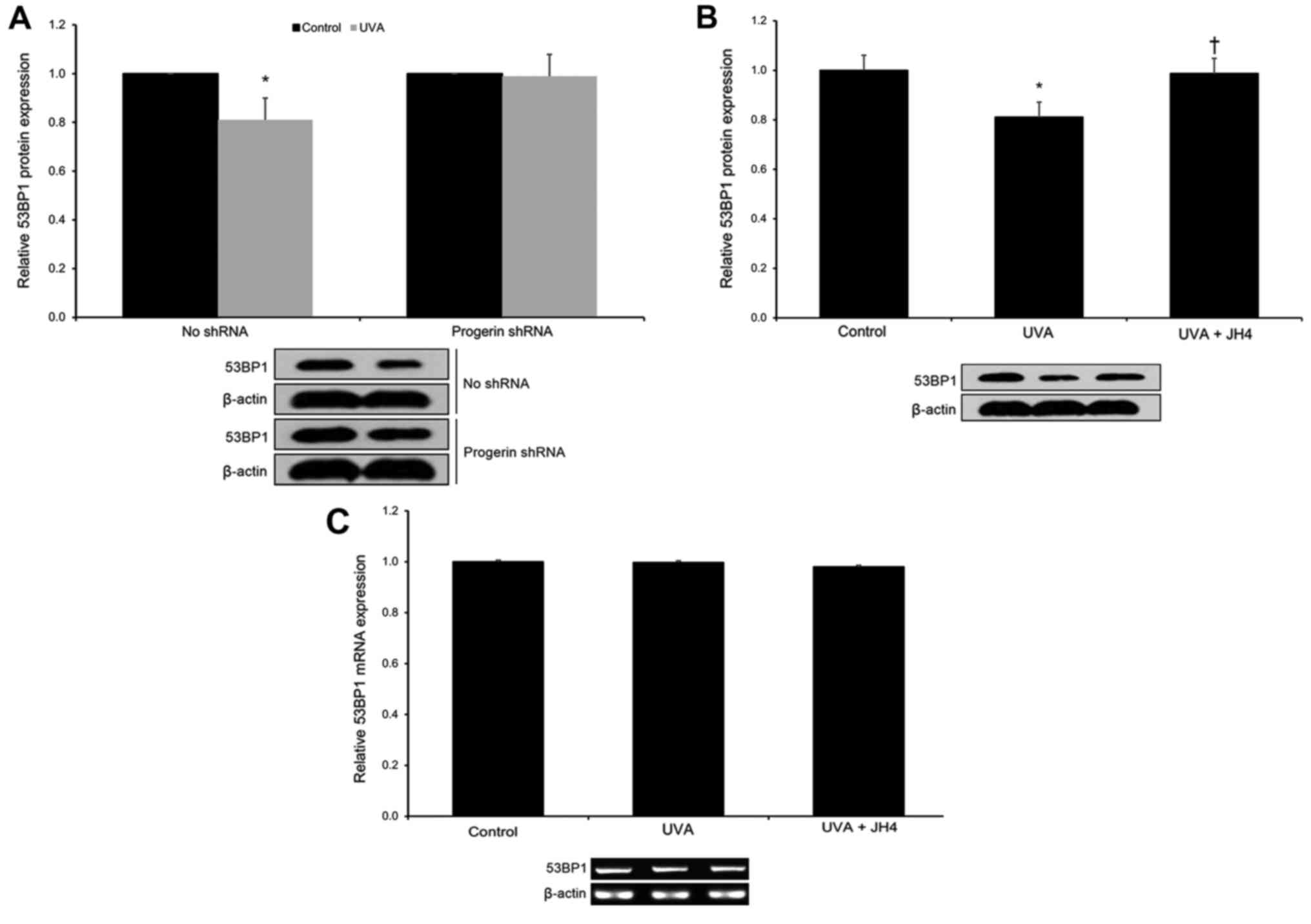
As lamin A has been shown to promote 53BP1 levels
(7), we next hypothesized that
UVA-induced progerin-lamin A complex formation (via the decrease of
free lamin A protein levels) may result in 53BP1 downregulation in
UVA-irradiated cultured human neonatal keratinocytes. First, we
found that silencing progerin (via a progerin-specific shRNA) was
able to rescue UVA-induced 53BP1 protein downregulation (p<0.05;
Fig. 4A). Next, we demonstrated
that blocking progerin-lamin A interaction by JH4 was able to
rescue UVA-induced 53BP1 protein downregulation (p<0.05;
Fig. 4B). As a control experiment,
we ascertained that JH4 administration produced no significant
effects on 53BP1 mRNA levels (p>0.05; Fig. 4C). These findings indicate that UVA
exposure significantly decreases 53BP1 protein levels in human
keratinocytes via enhanced progerin-lamin A complex formation.
As lamin A deficiency has been shown to inhibit NHEJ
DSB repair through the promotion of 53BP1 degradation (10), we next hypothesized that UVA-induced
progerin-lamin A complex formation may inhibit NHEJ DSB repair in
UVA-irradiated cultured human neonatal keratinocytes. In order to
test this hypothesis, we performed a set of neutral comet assays on
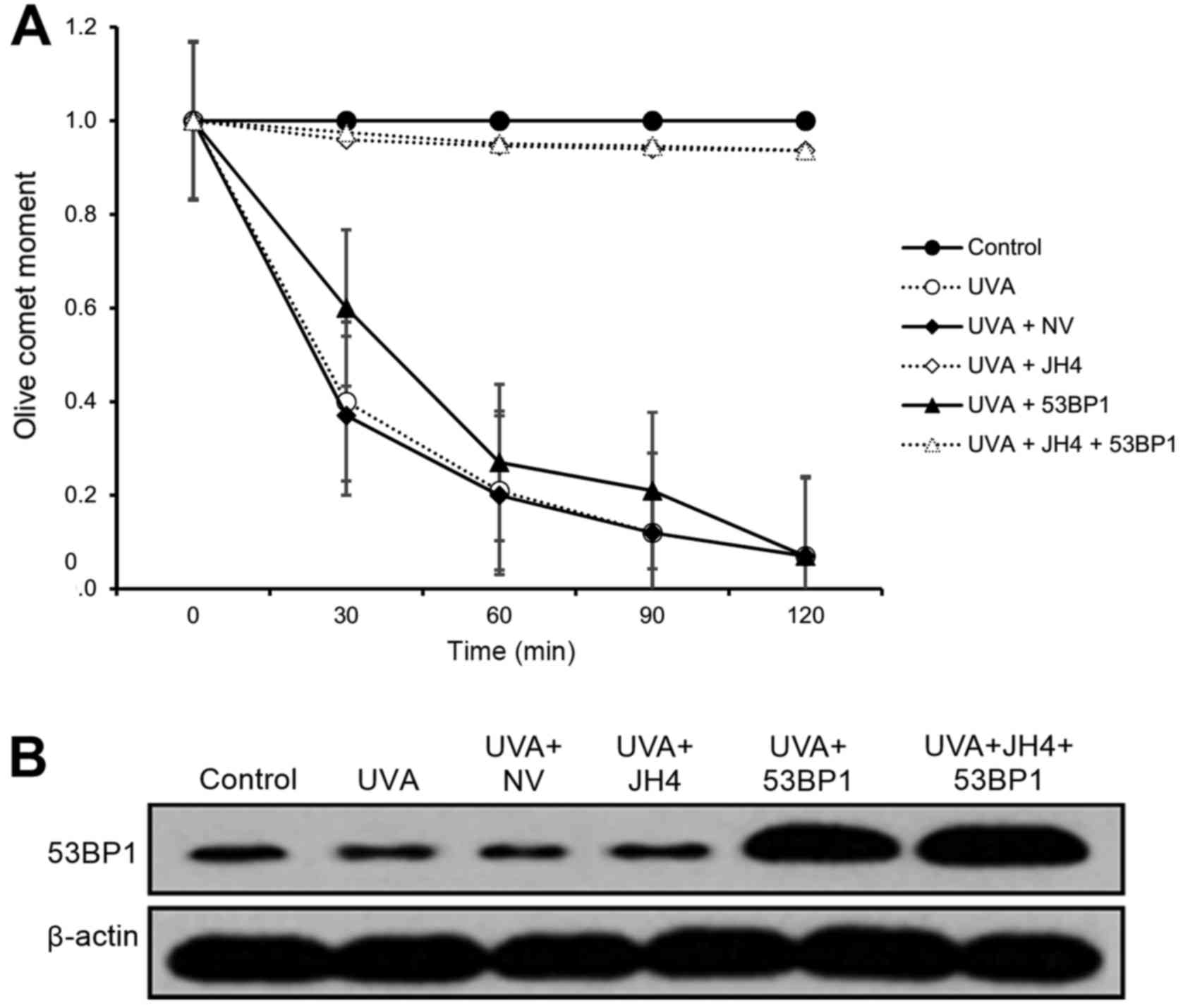
cultured human neonatal keratinocytes. As expected, we found that
UVA irradiation significantly decreased NHEJ DSB repair activity
(p<0.05; Fig. 5A). Notably, JH4
treatment was able to fully rescue NHEJ DSB repair activity
irrespective of 53BP1 overexpression (p<0.05; Fig. 5A). However, 53BP1 overexpression
alone was only able to partially rescue NHEJ DSB repair activity
(p<0.05; Fig. 5A). These
findings indicate that UVA-induced progerin-lamin A complex
formation is largely responsible for the inhibition of
53BP1-mediated NHEJ DSB repair activity.
Discussion
Progerin is a shortened, farnesylated splicing
variant of the LMNA gene, which has been most popularly associated
with the accelerated aging genetic disorder termed
Hutchinson-Gilford progeria syndrome (HGPS) (18). The progerin protein dysfunctionally
localizes on the inner surface of the nuclear membrane, where it
interferes with several critical lamin A-associated nuclear
processes such as chromatin organization, DNA damage response
pathways, gene transcription activity, and maintaining telomere
integrity (19). Notably, progerin
is expressed at low levels in healthy people and increases in
expression over the course of normal physiological aging (20); moreover, progerin upregulation has
also been associated with prostate cancer and renal cell carcinoma
(21). Thus, an improved
understanding of the role of progerin in aging and carcinogenesis
may be helpful in developing anti-aging and anticancer
therapeutics. Unfortunately, there has been limited research
regarding the role of progerin in photoaging and
photocarcinogenesis in skin keratinocytes. The present study is the
first to demonstrate that UVA-induced progerin upregulation
adversely affects 53BP1-mediated NHEJ DSB repair in human
keratinocytes via progerin-lamin A complex formation.
First, from a large population of human skin
samples, we discovered that progerin upregulation in human
keratinocytes is significantly associated with advancing age, not
BCC status. This finding concurs with a study by Miller et
al which showed increased fibroblastic progerin expression in
aged human patients and demonstrated that experimentally induced
progerin expression in induced pluripotent stem cell (iPSC)-derived
fibroblasts and neurons induces several aging-related
characteristics in vitro (19). Our findings also concur with those
of Olive et al which discovered progerin expression in
coronary arteries of aged human patients and demonstrated that
coronary arterial progerin expression significantly increases with
advancing age (22). Second, we
found that UVA exposure significantly upregulates progerin
expression in human keratinocytes by favoring alternative LMNA gene
transcript splicing. These findings concur with those of the
Firoozan et al and Takeuchi et al research groups,
which have shown that UVA radiation induces progerin upregulation
in human melanocytes and fibroblasts (11,23).
Moreover, they also demonstrated that this UVA-induced progerin
upregulation is mediated by radiation-driven oxidative damage,
which promotes alternative splicing of the LMNA gene transcript
(11). As these combined findings
show an association between age, UVA exposure and progerin
upregulation in human keratinocytes, but no significant association
between BCC status and progerin expression, our findings suggest
that progerin upregulation in human keratinocytes is associated
with photoaging, but not photocarcinogenesis.
A recently published study by Lee et al
revealed that lamin A is an important binding target of progerin
(12). Moreover, they also
demonstrated that the chemical compound JH4 selectively binds to
progerin, thereby blocking progerin-lamin A complex formation
(12). Therefore, we applied JH4 in
order to determine whether the effects of UVA on lamin A protein
levels are affected by progerin-lamin A complex formation. First,
we found that the silencing of progerin was able to rescue
UVA-induced free lamin A protein downregulation. Second, after
ascertaining that JH4 blocks the interaction of progerin and lamin
A in cultured human keratinocytes, we found that JH4 treatment was
able to rescue free lamin A protein expression in UVA-irradiated
keratinocytes. These findings demonstrate that UVA exposure
significantly decreases free lamin A protein levels in human
keratinocytes through the promotion of progerin-lamin A complex
formation.
Lamin A deficiency has been shown to decrease
nuclear accumulation of the DNA damage response protein 53BP1 at
UV-induced foci through the enhancement of 53BP1 degradation
(10). In the present study, we
demonstrated that UVA exposure significantly decreased 53BP1
protein levels in human keratinocytes via enhanced progerin-lamin A
complex formation (a process which decreased free lamin A levels).
However, we did not examine the precise mechanism(s) by which the
complex formation-driven decrease in free lamin A levels affects
53BP1 protein expression, as these have been reported elsewhere. A
previous study by Gonzalez-Suarez et al, demonstrated that
lamin A deficiency drives cysteine protease cathepsin L
(CTSL)-mediated degradation of the 53BP1 protein (24). Moreover, Gibbs-Seymour et al
demonstrated that lamin A directly binds to the 53BP1 Tudor domain
via a ataxia telangiectasia mutated (ATM)-mediated mechanism,
thereby promoting nuclear 53BP1 retention (7). As 53BP1 is a key effector of lamin A
(7), further research on the
downstream mechanism(s) involved in the effects of free lamin A on
53BP1 protein degradation and nuclear localization are still
needed.
Progerin has been shown to slow NHEJ DSB DNA repair,
which leads to heightened DSB accumulation over time (13). Specifically, progerin-overexpressing
cells display a significantly increased DSB event frequency via
NHEJ, with no significant effect observed upon DSB event frequency
via HR (13). Notably, in the
present study, JH4 treatment was able to fully rescue NHEJ DSB
repair activity irrespective of 53BP1 overexpression, while 8-fold
53BP1 overexpression alone was only able to partially rescue NHEJ
DSB repair activity. These combined findings suggest that
UVA-induced progerin-lamin A complex formation is primarily
responsible for 53BP1-mediated losses in NHEJ DSB repair activity,
but there are other non-53BP1-mediated mechanism(s) involved in
NHEJ DSB repair activity that are also adversely affected by
progerin-lamin A complex formation. Indeed, Ghosh et al have
reported that free lamin A protein recruits and facilitates sirtuin
6 (SIRT6)-mediated NHEJ DSB repair (25,26).
Thus, UVA-induced progerin-lamin A complex formation may adversely
affect NHEJ DSB repair through the attenuation of both 53BP1 and
SIRT6 pathways. As this field of investigation is relatively novel,
further studies are still needed to examine the downstream
effector(s) of lamin A that affect DSB repair activity.
In conclusion, the present study is the first to
demonstrate that UVA-induced progerin upregulation adversely
affects 53BP1-mediated NHEJ DSB repair in human keratinocytes via
progerin-lamin A complex formation. These findings improve our
understanding of the role of progerin in photoaging and may provide
additional insights to support the development of anti-aging skin
therapeutics.
Acknowledgements
The present study was supported by the Natural
Science Foundation of China (81573027).
References
|
1
|
Moan J, Grigalavicius M, Baturaite Z,
Dahlback A and Juzeniene A: The relationship between UV exposure
and incidence of skin cancer. Photodermatol Photoimmunol Photomed.
31:26–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iannacone MR, Hughes MCB and Green AC:
Effects of sunscreen on skin cancer and photoaging. Photodermatol
Photoimmunol Photomed. 30:55–61. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kabir Y, Seidel R, Mcknight B and Moy R:
DNA repair enzymes: An important role in skin cancer prevention and
reversal of photodamage - a review of the literature. J Drugs
Dermatol. 14:297–303. 2015.PubMed/NCBI
|
|
4
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schwertman P, Bekker-Jensen S and Mailand
N: Regulation of DNA double-strand break repair by ubiquitin and
ubiquitin-like modifiers. Nat Rev Mol Cell Biol. 17:379–394. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Surovtseva Y, Jairam V, Sundaram R, Bindra
R and Herzon S: A high-throughput, high-content assay for the
discovery of new inhibitors of DNA double-strand break repair.
Cancer Res. 76(Suppl 14): 21742016.doi:
10.1158/1538-7445.AM2016-2174. View Article : Google Scholar
|
|
7
|
Gibbs-Seymour I, Markiewicz E,
Bekker-Jensen S, Mailand N and Hutchison CJ: Lamin A/C-dependent
interaction with 53BP1 promotes cellular responses to DNA damage.
Aging Cell. 14:162–169. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Canny M, Wan L, Fradet-Turcotte A,
Orthwein A, Moatti N, Juang YC, Zhang W, Noordermeer SM, Wilson MD,
Vorobyov A, et al: A genetically encoded inhibitor of 53BP1 to 1
stimulate homology-based gene editing. bioRxiv.
0609542016.https://doi.org/10.1101/060954
|
|
9
|
Her SC and Her C: Targeting DNA
double-strand break repair in cancer therapy. J Mol Genet Med.
9:e1062015.
|
|
10
|
Redwood AB, Perkins SM, Vanderwaal RP,
Feng Z, Biehl KJ, Gonzalez-Suarez I, Morgado-Palacin L, Shi W, Sage
J, Roti-Roti JL, et al: A dual role for A-type lamins in DNA
double-strand break repair. Cell Cycle. 10:2549–2560. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takeuchi H and Rünger TM: Longwave UV
light induces the aging-associated progerin. J Invest Dermatol.
133:1857–1862. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee SJ, Jung YS, Yoon MH, Kang SM, Oh AY,
Lee JH, Jun SY, Woo TG, Chun HY, Kim SK, et al: Interruption of
progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria
syndrome phenotype. J Clin Invest. 126:3879–3893. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Waldman AS, Chowdhary S, Patrick A, Hersey
M and Waldman BC: The influence of progerin expression on the
nature of DNA double-strand break repair. The FASEB J. 30:(Suppl
1): S576.42016.
|
|
14
|
Mistry DS, Chen Y, Wang Y and Sen GL:
Transcriptional profiling of SNAI2 regulated genes in primary human
keratinocytes. Genom Data. 4:43–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Malaisse J, Pendaries V, Hontoir F, De
Glas V, van Vlaender D, Simon M, de Rouvroit C Lambert, Poumay Y
and Flamion B: Hyaluronan does not regulate human epidermal
keratinocyte proliferation and differentiation. J Biol Chem.
291:6347–6358. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang H, Xiong Z-M and Cao K: Mechanisms
controlling the smooth muscle cell death in progeria via
down-regulation of poly(ADP-ribose) polymerase 1. Proc Natl Acad
Sci USA. 111:pp. E2261–E2270. 2014; View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Woods M, Pant R and Mallya SM: Cyclin D1
and cyclin D-dependent kinases enhance oral keratinocyte
proliferation but do not block keratinocyte differentiation. Int J
Oncol. 37:1471–1475. 2010.PubMed/NCBI
|
|
18
|
Musich PR and Zou Y: DNA-damage
accumulation and replicative arrest in Hutchinson-Gilford progeria
syndrome. Biochem Soc Trans. 39:1764–1769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miller JD, Ganat YM, Kishinevsky S, Bowman
RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, et al: Human
iPSC-based modeling of late-onset disease via progerin-induced
aging. Cell Stem Cell. 13:691–705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kubben N, Zhang W, Wang L, Voss TC, Yang
J, Qu J, Liu GH and Misteli T: Repression of the antioxidant NRF2
pathway in premature aging. Cell. 165:1361–1374. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kennedy BK: A new connection between VHL
and cancer threads through progerin. Cell Cycle. 12:2721–2722.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Olive M, Harten I, Mitchell R, Beers JK,
Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, et al:
Cardiovascular pathology in Hutchinson-Gilford progeria:
Correlation with the vascular pathology of aging. Arterioscler
Thromb Vasc Biol. 30:2301–2309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Firoozan S, Takeuchi H and Ruenger T:
Longwave ultraviolet light induces the aging-associated progerin in
melanocytes. Am J Med Genet. 2603:26242006.
|
|
24
|
Gonzalez-Suarez I, Redwood AB, Grotsky DA,
Neumann MA, Cheng EHY, Stewart CL, Dusso A and Gonzalo S: A new
pathway that regulates 53BP1 stability implicates cathepsin L and
vitamin D in DNA repair. EMBO J. 30:3383–3396. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ghosh S, Liu B, Wang Y, Hao Q and Zhou Z:
Lamin A is an endogenous SIRT6 activator and promotes
SIRT6-mediated DNA repair. Cell Reports. 13:1396–1406. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mao Z, Hine C, Tian X, van Meter M, Au M,
Vaidya A, Seluanov A and Gorbunova V: SIRT6 promotes DNA repair
under stress by activating PARP1. Science. 332:1443–1446. 2011.
View Article : Google Scholar : PubMed/NCBI
|