Introduction
When blood supply to a solid tumor is limited, such
as by interrupted or abnormal vasculature, a hypoxic
microenvironment is generated (1).
Effects of the hypoxic state, and the induced physiological and
tumorigenic changes, have been extensively studied (2–4) and
include cell metastasis (5),
neovascularization (6), cell
invasiveness (7) and resistance to
radiotherapy, and chemotherapy (8–10). The
various regulatory factors contributing to hypoxia-associated
signaling and mechanisms have been proposed as targets for oncology
research (11), and extensive
proteomic studies of hypoxia in many types of tumor cells have been
carried out and reported in recent years.
The concept of ‘proteome’ was first publicly
proposed in 1994, and was described as the full profile of proteins
expressed by all genes in the genome of a cell or the particular
profile of proteins expressed by a cell at a certain phase
(12). Since then, proteomic study
has been applied to many fields of biological research (13–15).
Development of the mass spectrometry (MS) technique significantly
advanced our ability to identify proteins and now represents the
main research method of proteomic analysis (16–18).
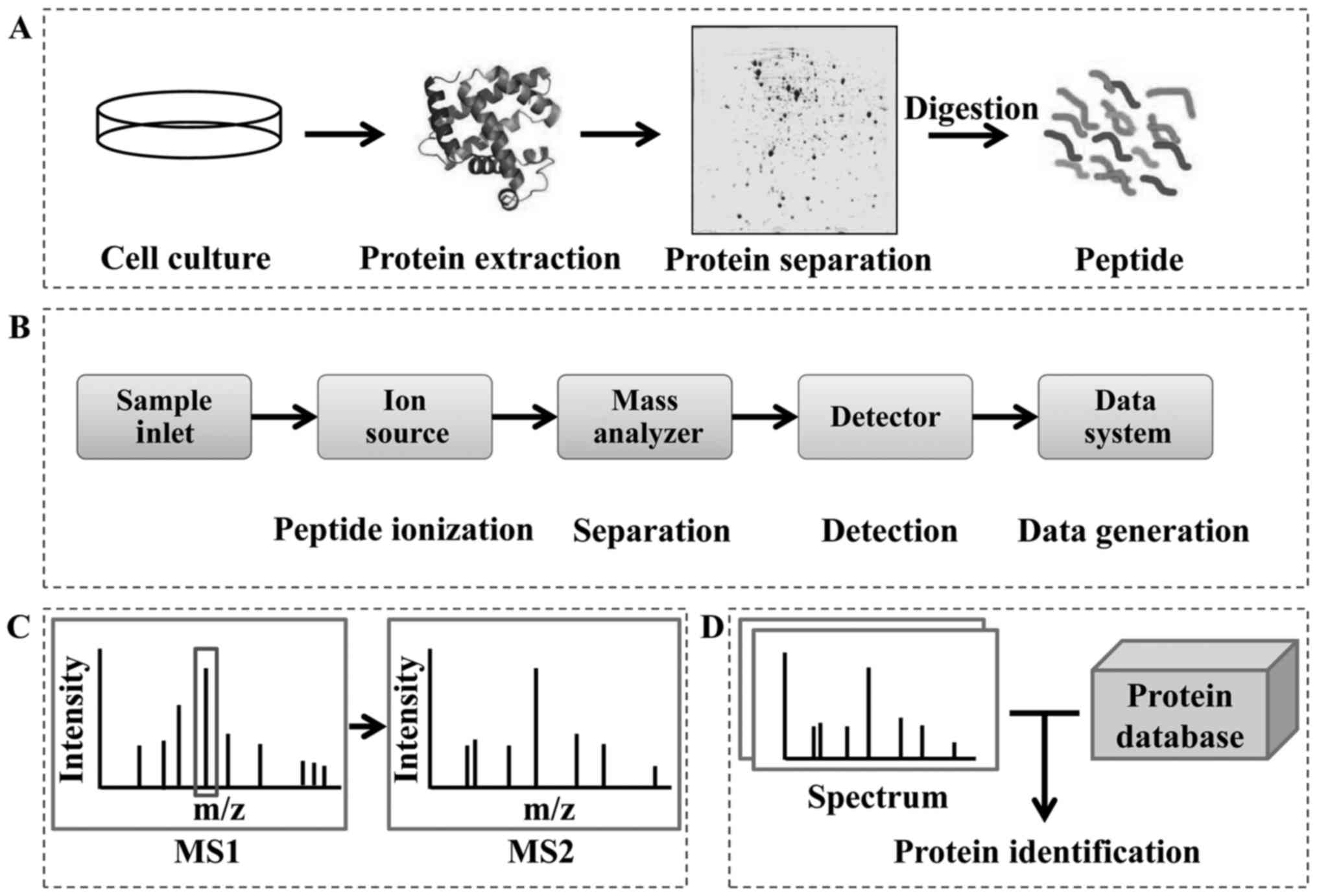
MS-based ‘shotgun’ proteomics is the prototype method and is
currently in wide use (Fig. 1). The
mass spectrometer instrument consists of a sample inlet, ion
source, mass analyzer, detector and data system (Fig. 1B). After passage through the sample
inlet, peptides are ionized in the ion source chamber and separated
in the mass analyzer chamber. The mass-to-charge ratio (m/z) of the
peptides is determined by the detector. Finally, the identity of
the proteins is confirmed by bioinformatic analysis, which
frequently involves automated searching of protein databases
(Fig. 1C and D).
Protein samples are usually prepared for MS
detection by gel electrophoresis separation (or resolution by other
methods) and digestion (Fig. 1A).
The most widely used gel-dependent protein separation technique is
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE), which was established in 1967 by Shapiro et al
(19,20) and was improved by Weber and Osborn
in 1969 (21). SDS, an anion
detergent, serves to break the hydrogen bonds between molecules,
causing breakdown of the protein structure. Protein migration rates
in gel electrophoresis depend on the molecular weight (MW), which
itself is affected by the concentration of the gel. Another
technique for separating proteins is two-dimensional gel
electrophoresis (2DE), which was established in 1975 (22). In this method, proteins are firstly
separated by their respective isoelectric points (pIs) and
secondly, in the perpendicular direction, by their respective MWs,
ultimately distributing throughout the gel as pI/MW-dependent
protein spots. On the basis of 2DE, 2D-difference in gel
electrophoresis (DIGE) was introduced by Unlü et al
(23). This expanded method relies
on protein labeling with fluorescent dye labeling to improve the
accuracy of quantitation (24).
Different proteins can then be detected by the MS technique and
finally identified by bioinformatics. Since 2DE allows for the
properties of proteins to be visualized (i.e. pI, MW and relative
abundance), it has emerged as a key tool for comparative proteomic
studies. However, the experimental operations for 2DE are not
automated, the separated proteins cannot be accurately quantified
and the proteins with extreme pIs or insolubility are not separated
well by 2DE.
To address the above concerns, quantitative
proteomic techniques have been developed, representing two
categories segregated by the use of either a stable or unstable
isotope for labeling of the protein samples. In label-free
quantification, the quantity can be determined based upon the
extracted ion current (XIC) (25)
or using the method of spectral counting (SC) (26). The usual stable isotopic labeling
quantification techniques, however, include the stable isotope
labeling by amino acids in cell culture (SILAC) (27), isotope-coded protein labels
(28), isobaric tag for relative
and absolute quantitation (iTRAQ) (29) and tandem mass tag (30). The same peptides with different
labels are presented in the same spectrogram as isotopic peaks with
fixed difference in MW. In label-free quantification, sample
preparation is simple (no label is required) and the cost is
relatively low, but the quantitative efficiency depends on the
stability of the mass spectrometer instrument itself. The
quantitative proteomic techniques using an unstable (radioactive)
isotope label is very specific and provides direct detection, but
the well-known safety issues are a disadvantage. In contrast,
stable isotope labels usually have high sensitivity and high
quantitative accuracy, but the operation is complicated and
features a relatively high cost.
Proteomic techniques make it possible to gain
greater insight into hypoxia by improving our ability to analyze
the underlying molecular mechanisms and consequences of the hypoxic
condition through protein expression. Herein, we describe, for the
first time, the collective applications of MS-based proteomic
analysis for assessing tumorous microenvironment hypoxia. Various
types of tumors have been subject to the study of hypoxia effects
and a multitude of proteins affected by the hypoxic condition have
been identified. Several proteins have been found repeatedly across
the studies of different tumor types, suggesting their potential
roles in general hypoxic regulation. In addition, proteins have
been identified that are distinctive to specific tumor types, and
as such represent possible biomarkers. A better understanding of
the hypoxia factors that compose the dynamic tumor proteome may
help advance both our overall understanding of tumorigenesis and
tumor cell survival, as well as our ability to generate innovative
antitumor therapies.
2DE/2D-DIGE-coupled MS
The 2DE technique has been widely used in
comparative proteomic analysis. For this, protein samples
representing different experimental conditions are separated on
respective gels (as detailed above) thus, that the relative
expression level, pI or MW changes visualized in one gel can be
compared to the other(s) for the different conditions being
assessed. The differentially expressed proteins can then be
selectively identified by coupled MS detection. The more newly
developed 2D-DIGE technique, in contrast, provides more accuracy in
finding differentially expressed proteins, due to the fluorescent
dyes used to label the various samples. Both of these methods,
however, have successfully been applied to the protein samples of
various tumor types, including head neck squamous cell carcinoma
(HNSCC), cervix carcinoma (i.e. HeLa cells), pancreatic ductal
adenocarcinoma (PDAC), leukemia and breast cancer.
HNSCC
Chen et al (31) reported a proteomic study of
hypoxia-regulated proteins in HNSCC in 2004. In total, more than
1,000 protein spots were separated in gels under hypoxic and
normoxic conditions. Twenty of those proteins showed increased
expression (>1.5-fold) during hypoxia. Among these, the IκB
kinase β (IKKβ) and MKK3b are known to be highly expressed in
cancer, and the density-regulated protein 1, P150glued,
nuclear transport factor 2, binder of ARL 2, paxillin and
transcription termination factor I represented newfound
hypoxia-inducible factors. Further analysis of IKKβ in hypoxia
revealed that it is a regulator of the nuclear factor-κB (NF-κB)
transcription factor under hypoxia conditions, which was in
accordance with previous studies (32). In patients with HNSCC, this enzyme
mediates cell survival during hypoxia and the expression correlates
with tumor oxygenation, suggesting that it may represent a novel
endogenous tumor marker.
Cervix carcinoma
In 2007, Magagnin et al (33) reported on protein expression changes
in HeLa cells under hypoxic and reoxygenation conditions. Seven of
14 differentially expressed protein spots for the reoxygenation
samples were identified by electrospray ionization
(ESI)-QTRAP® MS (AB SCIEX, Framingham, MA, USA) and were
found to be associated with cellular processes and stress response
pathways. These proteins included VCP/p97, FUSE binding proteins 2
and 3 (FBP2/FBP3) and ribosomal protein P0 (RPP0). The VCP/p97
protein is a hexameric ATPase and it is associated with various
processes of cellular regulation (34). The FBP2 and FBP3 proteins are
associated with RNA processing and have been demonstrated as being
involved in the regulation of protein expression in response to
hypoxia (35,36) and other conditions, such as
irradiation (37) and oncogenic
signaling (38). Overall, these
identified proteins play roles in the resistance of tumor cells to
hypoxic exposure. Magagnin et al (39) further reported on a study of
proteome changes in HeLa cells in 2008. Several proteins were found
to be regulated by mammalian target of rapamycin (mTOR) and its
downstream effector 4E-BP1, both under hypoxic and normoxic
conditions. These proteins had been previously reported as
associated with tumor cell motility, invasion and metastasis, all
of which contribute to the adverse tumor phenotype (40–43).
PDAC
A study of hypoxia-regulated proteins in PDAC was
reported by Cui et al (44)
in 2009. Thirty-five unique expressed proteins were identified in
PDAC, as compared to proteins in pancreatic ducts; these included
21 upregulated and 14 downregulated proteins. Seven of the 21
increased proteins had been previously reported to be regulated by
hypoxia (45–50), and these included Annexin A5, fatty
acid synthase, fructose bisphosphatase aldolase A,
glucose-regulated protein 78 (Grp78), macrophage migration
inhibitory factor (MIF), phosphoglycerate kinase-1 (PGK-1) and
transketolase.
Leukemia
Liao et al (51) reported a study of different leukemic
cell lines, in which they aimed to identify direct targets of
hypoxia-induced factor (HIF)-1 and provide new insights into the
molecular mechanisms associated with HIF-1α-mediated tumor
metastasis. According to the results, Annexin A1, CapG and S100A4
genes were significantly induced by hypoxia in 5 of the adherent
cell lines tested as well as in the non-adherent U937 cells, and
the expression could be blocked by silencing the HIF-1α gene. CapG
is a member of the gelsolin superfamily and it plays a role in the
regulation of actin filament length by severing filaments (52). S100A4 is involved in cancer cell
migration and invasion (53,54)
and the functions were identified in the present study. Ultimately,
S100A4 and CapG have been suggested as novel direct target
candidates of HIF-1α.
Breast and colon cancers
Bartkowiak et al (55) reported in 2010 their results of a
study of the breast cancer disseminated tumor cell (DTC) line,
which is established from bone marrow, a known hypoxic
microenvironment. The unfolded protein response (UPR) proteins
Grp78 and Grp94 and the protein disulfide-isomerase were identified
as overexpressed. These UPR proteins could be applicable as novel
biomarker proteins for DTC detection in cancer patients. A few
years later, in 2013, Grandjean et al (56) identified a potential colon cancer
biomarker through their research of the human colorectal carcinoma
HCT116 and HT29 cell lines, through which the phosphorylated form
of translation elongation 2 was verified as a new tumor-associated
antigen.
As a traditional proteomic technique, 2DE coupled MS
has been broadly applied in proteomic studies. However, the 2DE
protein separation step is not high-throughput and the overall
operation is complicated. Moreover, the strategy is not highly
discriminative, limiting accuracy of identification of low
abundance proteins. Quantitative proteomics were developed to
surmount these limitations, and their advantages over the 2DE-based
methods have been proven over recent years.
Quantitative MS
Label-free quantitative MS
A multitude of label-free quantitative proteomic
studies have been carried out in recent years. The simple sample
preparation process and lack of label make label-free quantitation
easy to perform. However, the accuracy of this method depends
greatly on the stability of the mass spectrometer instrument used.
To date, tumor cells from gliomas, hepatocellular carcinoma (HCC)
and colorectal cancer as well as human umbilical vein endothelial
cells (HUVECs) have all been studied by label-free quantitative MS
techniques.
Glioma
Gliomas (brain tumors) have a general poor
prognosis. This tumor type was studied by the proteomic approach by
Yoon et al (57) and
reported on in 2014. The exosome and soluble fractions were
isolated from the U373MG glioma cell line under hypoxic and
normoxic conditions and analyzed; a total of 239 proteins were
identified. The upregulated proteins were identified based on a
label-free quantitative analysis. Vascular endothelial growth
factor, stanniocalcins 1 and 2 (STC1 and STC2, respectively), and
insulin-like growth factor binding proteins 3 and 6 (IGFBP3 and
IGFBP6, respectively) were found to be enriched in the soluble
fraction, and lysyl oxidase homolog 2 was found to be enriched in
the exosomal fraction. Among these proteins, the STCs and IGFBPs
are secretory proteins enriched under hypoxia conditions. Previous
studies in the literature have revealed that STC1 is a calcium- and
phosphate-regulating hormone in the blood of fish, and the
homologues STC1 and STC2 were also subsequently identified in
mammals (58,59). The expression level of the STCs
correlated with glioma grade in the present study, and they were
also found to be related with hypoxia-dependent migration of tumor
cells. Thus, the STCs represent potential biomarkers for cancer
diagnosis, as was purported by other previous studies (60,61).
HCC
Malignancy progression in hypoxia of HCC was studied
by a label-free quantitative proteomic strategy (62). A single significantly increased
protein was identified: prohibitin 2 (PHB2). Previous studies have
demonstrated PHB2 to be associated with the mitochondria, in
various aspects (63–66). When the PHB2 expression was
inhibited or the HCC cells were treated with interfering RNAs
against PHB2, the cells showed changes in phenotype of adaption
ability to hypoxic microenvironment, in resistance to
chemotherapy-induced apoptosis and in cell growth. These results
suggested that the PHB2 protein plays important roles in HCC
malignancy progression, providing a reference for treatments based
on this protein.
Colorectal cancer
The HIF-1α gene in HCT116 colorectal cancer cells
was analyzed by a label-free quantitative method, with the aim of
studying the metabolic pathways in lipogenesis (67). In total, 3,632 proteins were
detected and identified. The results revealed that the lipid
metabolites are regulated by hypoxia through both HIF1α-dependent
and -independent pathways. The HIF1α-dependent pathways include
enzymatic steps in fatty acid synthesis and the Kennedy pathway.
The HIF1α-independent pathways include palmitate, stearate, PLD3
and PAFC16 pathways.
HUVECs
Considering the necessity of vessel formation in
tumor growth, invasion and metastasis, a label-free quantitative
proteomic study was conducted using HUVECs under hypoxic
conditions. Twenty-seven sialoglycoproteins on the surface of
HUVECs were identified as overexpressed (68), in addition to the CD105,
neuropilin-1 and CLEC14A proteins that had been previously
described by other studies of hypoxia (69–71).
Various new proteins were identified as hypoxia regulated,
including glucose transporter SLC2A1 (GLUT-1), TMEM16F and stromal
cell-derived factor (SDF) 4.
Stable isotope-based quantitative
MS
In the SILAC technique, cells are labeled by
isotopic amino acid during the culture process. This is an
efficient and sensitive method, but the cost of the label and the
extensive time required for labeling limit its suitability for all
protein samples, particularly those from tissue or plasma. The
iTRAQ technique is also effective and is sufficiently applicable to
various types of protein samples. Other isotope-based labeling
methods, such as those using 15N and 18O,
have also been successfully applied in proteomic studies. To date,
these collective quantitative MS-based techniques have been used in
studies of melanoma, neuroblastoma, mammary cancer, von
Hippel-Lindau (VHL) tumor, epidermoid carcinoma, lung cancer and
adenocarcinoma.
Melanoma
In 2006, a study of the plasma membrane of the
hypoxia-adapted murine B16F10 melanoma cells was reported, in which
the quantitative proteomic study was based on
16O/18O stable isotopic labeling and
multidimensional liquid chromatography-tandem MS (72). In total, 24,853 peptides were
identified, corresponding to 2,433 proteins. Under hypoxic
conditions, aminopeptidase N (CD13), carbonic anhydrase IX,
potassium-transporting ATPase, matrix metalloproteinase 9 and SDF1
were upregulated. Moreover, CD13 and SDF1 were validated in human
melanoma cell lines, confirming the increased expression during
hypoxia. Identification and characterization of novel
hypoxia-induced membrane proteins may almost certainly help in the
research efforts aimed at the treatment of melanoma.
Neuroblastoma
Small ubiquitin-like modifier (SUMOP2/3)
conjugation, which is usually a consequence of transient cerebral
ischemia, was studied using SILAC-based quantitative proteomics in
2012 (73). The neuroblastoma B35
cell line was exposed to the ischemic condition and the changes in
levels of specific SUMOylated proteins were assessed using a stable
isotope labeling method. Lysates were mixed equally with control
proteins and analyzed by 1D SDS-PAGE combined with LC-MS/MS. In
total, 118 putative SUMO3-conjugated proteins were identified, of
which 22 showed changes in expression that were significant. The
protein-conjugated SUMO1/2 and ubiquitin were identified as
upregulated and the OGD-induced ubiquitination could be completely
blocked by gene silencing of SUMO2/3. Ultimately, the present study
demonstrated an association between the SUMO and ubiquitin
conjugation pathways.
Mammary cancer
A comprehensive hypoxic quantitative proteomic study
was conducted using matrix-assisted laser desorption/ionization MS
imaging (commonly known as MALDI-MSI) (74). SILAC-labeled mammary cancer cells
were studied and changes in the proteome under hypoxic conditions
were identified. At the same time, laser-capture microdissected
samples isolated from normoxic and hypoxic regions of mammary
tumors were detected by quantitative MS, and the localization of
hypoxia-regulated proteins was also analyzed. In the first part of
the study, a total of 1,416 proteins were identified, among which
131 had alterations in expression, including 60 that were
upregulated and 71 that were downregulated. In the second part of
the study, 164 and 118 proteins were identified in hypoxia and
normoxic regions, respectively, and 89 proteins were found to exist
in both the hypoxia and normoxic regions.
VHL
The VHL tumor was studied using clear-cell-type
renal cell carcinoma (RCC) samples (75). In the present study, the proteomic
and phospho-proteomic analyses were combined to analyze 786-O RCC
(±VHL) cells in order to identify VHL-associated hypoxic responses.
However, the upregulated GLUT-1 (76,77)
and N-myc downstream regulated gene 1 (NDRG1) (78), which are known biomarkers of RCC,
downregulated intracellular carbonic anhydrase II (CA2) was also
identified. CA2 was demonstrated to govern cellular response to the
CO2 fluctuations that accompany hypoxia in tumors.
Epidermoid carcinoma
iTRAQ-based quantitative proteomics have been
applied to determine the protein expression alterations driven by
hypoxia in various compartments of the A431 human epithelial
carcinoma cell line. In 2012, a study of iTRAQ-based quantitative
proteomic study of global protein expression and functional changes
in A431 carcinoma cells under hypoxic and reoxygenated conditions
was published (79). A total of
4,316 proteins were identified, among which more than 1,200
proteins showed changes in their expression. Hypoxia and
reoxygenated conditions affected various pathways, including DNA
repair, glycolysis, integrin, glycoprotein turnover and STAT1
signaling. Ultimately, the upregulation of glycolysis, integrin,
glycoprotein synthesis and downregulation of STAT1 pathways
increased metastatic activity of the A431 cells. The non-homologous
end-joining pathway (NHEJ) was also identified as upregulated,
representing a novel finding at the time. The NHEJ pathway plays an
important role in DNA repair of irradiated cells, and its
upregulation under the hypoxic condition may enhance the
radioresistance of tumor cells (80).
In addition to the total hypoxia-mediated protein
profile identified in A431 cells, profiles of the secreted and the
exosome proteins, the integrin family of glycoproteins and the
chromatin-binding proteins has been the subject of recent
investigations using the iTRAQ-based quantitative proteomic
strategy. In 2010, Park et al (81) reported that the secreted protein
profile and exosome profile in A431 cells were beneficial for tumor
cells to enhance angiogenic and metastatic potential. Different
hypoxic and reoxygenated exposure durations were applied to the
tumor cells, and the proteome changes were compared with cells
under normoxic condition. The quantitative proteomic analysis
revealed that the panel of secreted proteins was involved in
angiogenesis, focal adhesion, extracellular matrix-receptor
interaction and immune cell recruitment, all of which were
consistent with the enhanced angiogenic and metastatic potential
behaviors observed under hypoxic or reoxygenated conditions, such
as reduced cell adhesion, increased invasiveness and production of
a secreted protein panel supporting increased chorioallantoic
membrane angiogenic activity. The identified secreted and
exosome-associated protein panels suggested that modulation of the
microenvironment in order to facilitate angiogenesis and metastasis
could be induced in tumor cells by hypoxia.
In 2014, using the same iTRAQ-based quantitative
proteomic technique in A431 cells, Ren et al (82) reported that the N-glycosylation
modifications of integrin α3 (ITGA3) were inhibited by hypoxia and
that the hypoxia-induced ITGA3 glycosylation alterations prevented
its translocation to the plasma membrane. In this manner,
hypoxia-induced decrease of ITGA3 increased the invasive ability of
cancer cells. In that same year, Dutta et al (83) tested the hypothesis that
hypoxia-driven evolution of the chromatome promotes malignant
changes and development of therapy resistance in tumor cells; the
study used isolated chromatins from A431 tumor cells treated with
varying conditions of normoxia, hypoxia and re-oxygenation followed
by partial digestion with DNase I. The iTRAQ-based quantitative
proteomic analysis of changes in euchromatin- and
heterochromatin-associated proteins identified a total of 1,446
proteins, among which 819 proteins were found to have an alteration
in the topology under hypoxia. A novel chromatin organizer protein,
HP1HP3, was identified that served to mediate chromatin
condensation and lead to increased viability, radioresistance and
chemoresistance, and self-renewal. Thus, the effects of
hypoxia-induced chromatin protein changes on tumor behavior were
shown through a proteomic approach and a candidate therapeutic
target was identified.
Lung cancer
The effect of multiple stressors on human lung
cancer cells has been studied using the proteomic approach
(84). In particular, iTRAQ-based
quantitative proteomic analysis was applied to lung cancer cells,
and bioinformatic analysis revealed that a novel cancer invasion
and metastasis-related gene, CIM, was involved in both hypoxic
stress- and endoplasmic reticulum stress-associated pathways. These
results offer a reference for tolerance against multiple cellular
stresses to be evoked by CIM, which plays a role in promoting
metastatic cell survival.
Adenocarcinoma
A tumor structure study was carried out with a
proteomic approach in 2012 (85).
Unlike other studies, the present study divided the solid tumor of
interest into four regions: surface (SL), intermediate region (IR),
perinecrotic region (PN) and necrotic core (NC). The PN contained
non-proliferating hypoxic cells. Protein samples from the four
different regions were labeled using the iTRAQ technique and
analyzed by quantitative proteomics. In total, 887 proteins were
identified, of which 209 were upregulated and 114 were
downregulated in PN and NC compared to SL. Moreover, proteins with
significant changes in expression were detected in the
non-proliferating fraction. The present study offered candidate
targets that may be suitable for therapeutic intervention.
Advantages and disadvantages of various
MS-based proteomics in tumor hypoxia
Diverse MS-based methods have been applied to
tumorous hypoxia research, representing three categories of
methods: 2DE/2D-DIGE coupled MS, label-free quantitative MS and
stable isotope-based quantitative MS (Table I). The 2DE-based MS technique
combines 2DE and MS detection, with the identity rate relying on
the discriminatory capacity of the gel electrophoresis. Proteins
separated by 2DE/2D-DIGE, present in the gel as protein spots,
distribute in the relatively same position regardless of which
sample is run (i.e. normoxia, hypoxia). Different expression levels
can be visually indicated by differences in the size of a protein
spot or the grayscale image of the spot in the gel. The proteins of
interest can then be cut out and subjected to MS for
identification. The MW and pI of protein spots can be readily
determined since the distribution pattern is according to these two
features. However, the disadvantages of this technique are related
to these very aspects; the operation itself is complex and time
consuming and the reliance on pI and MW for the protein separation
leads to poor distinguishment of proteins with extreme pIs or
similar MWs.
 | Table I.Common proteomic techniques. |
Table I.
Common proteomic techniques.
| Technique | Description | Advantage | Limitation | Application in
hypoxia (Refs.) |
|---|
| 2DE/2D-DIGE-MS
coupled | Proteins are
separated by pIs and MWs, distributed throughout the gel as protein
spots | Information on
abundance, pIs and MWs are presented visually, and differentially
expressed proteins are identifiable by the coupled MS | Complex operation
that is not automated Proteins with extreme pIs or insolubility are
not separated well | (31,33,44,51,55,56) |
| Label-free
quantitative MS | Protein quantity is
confirmed by the extracted ion current or spectral counting | Sample treatment is
simple (no label is required) and the cost is relatively low
Protein information is attained without manipulation (i.e. the
labeling) | Quantitative
efficiency depends on the stability of the mass spectrometer
Repeated experiments are needed | (57,62,67,68) |
| Stable
isotope-based quantitative MS | Proteins are
isotope-labeled based on metabolic or chemical technique | High-throughput,
high sensitivity and high quantitative accuracy | Cost is relatively
high The different labeling techniques have their own
limitations | (72–75,79,81–85) |
The label-free quantitative MS technique has been
used extensively in more recent years. The XIC-based label-free
quantitative technique mainly relies on the m/z and the signal
intensity derived from the spectrogram. Changes in the
chromatographic peak in each m/z reflect changes in retention time
and signal intensity, which reflect the abundance differences that
occur in response to the various conditions under evaluation. In
practice, however, stability and reliability of an experimental
platform needs to be considered, and in this case retention time
should be adjusted to ensure alignment for the co-quantitative
peptides in different samples. The two most usual methods for
alignment are based on characteristic peaks before the database
searching is initiated or on identification results after the
database searching has been completed.
When assessing the individual quantitative peptides
present in each sample, a cross-search algorithm is used to predict
the retention time in other samples, which helps to reconfigure the
XIC. Quantification results of the corresponding peptides present
in each sample can be confirmed by SC-based quantitative technique,
which relies on the abundance of peptides generated by protein
digestion. A protein with high expression level generates more
peptides. In practice, the SC-based method is limited by
experimental error during its application in analysis. In general,
the XIC-based label-free quantitative technique has more accuracy,
sensitivity and repeatability than the SC-based methods,
particularly for the identification of low abundance proteins.
However, when these two methods are combined a new set of problems
arise.
Stable isotopic labeling quantitative proteomic
techniques mainly depend on the uses of isotopic label for the
protein sample of interest. The same peptides with different labels
may present in a single spectrogram as isotopic peaks with fixed
mass differences, which may help identify the abundance of these
peptides. Both the precursor ion and the fragment ion can be
labeled by stable isotope. Precursor ion labeling is classified as
either metabolic labeling or chemical labeling. SILAC is a usual
metabolic labeling technique, and essential amino acids, such as
arginine and lysine (27), labeled
with stable isotope are usually used for cell culture experiments.
Fragment ion labeling can provide ions with mass differences in
MS/MS fragmentation. iTRAQ has emerged as the most widely used
method for fragment ion labeling. Four or eight samples can be
labeled simultaneously by iTRAQ technique. Fragment ion labeling
has advantages in repeatability and convenience for result
calculation, which unfortunately can be affected by co-eluted
peptides. When the two procedures are compared, the precursor ion
labeling has very high efficiency but is very complicated and time
consuming.
Conclusion and perspectives
Cells in living organisms express particular panels
of proteins whose functionality maintains life and provides
response to disease or injury. Similarly, proteins expressed by
tumorous cells provide a systematic picture of the cell metabolic
activity, represented by the proteome. Proteomic changes under the
tumorous hypoxic microenvironment, as compared to the normoxic
state, reflect the cell regulation processes involved in tumor
development and persistence. MS-based proteomics has been
extensively applied in tumor hypoxia studies during the past
several years (Table I), allowing
for investigation of various tumor cells under hypoxic conditions
in order to search for response-related proteins that provide
further insights into the underlying molecular mechanisms and
identification of candidate biomarkers for clinical use. The more
recent introduction of high-throughput MS technologies has
facilitated protein detection on a large scale, and a plethora of
proteins involved in the hypoxic response have been identified. The
inherent differences among these collective studies, such as in
their study designs, as well as the internal differences of the
tumor cells themselves have resulted in an array of
hypoxia-associated proteins, the profiles of which include those
related to general hypoxia regulation and those related to specific
tumor types.
A series of novel biomarker proteins have also been
found by these proteomic studies, including the IKKβ in HNSCC
(31), the UPR proteins (Grp78,
Grp94 and protein disulfide-isomerase) in DTC (55), and GLUT1 and NDRG1 in RCC (75). The proteins common among the
different tumor cell research studies of hypoxic conditions (i.e.
likely regulator of the general or less specific, hypoxic response)
include the Grp78 protein that was identified in both PDAC
(44) and DTC (55), and GLUT-1 that was identified in
HUVECs (68) and RCC (75). The overall proteins with
differential expression under hypoxic conditions function in a wide
array of cancer-related activities with outcome implications,
including cell adaptation, migration and invasion, which is in
accordance with the radioresistance and chemoresistance effects of
hypoxia in tumor cells.
In view of the characteristics of the different
methods available for proteomic analysis, the 2DE-based techniques
have yielded relatively less proteins than the high-throughput
techniques, with the different expression levels being confirmed by
comparison of protein spots in gel. As to the MS-based quantitative
proteomic studies, the different expression levels have been
usually confirmed by bioinformatic analysis of datasets, adding
further power to the potential of this approach to identify a
greater number of proteins. Nonetheless, the proteins that have
been identified to date warrant further research to gain a greater
understanding of their functions or relations to metabolic
pathways. Future studies, involving other experimental techniques
such as metabolomic analysis, should be carried out to keep the
progress moving forward on the tumor hypoxia microenvironment.
The 2DE-based separation relies on the pI and MW,
which means that proteins with extreme pIs may not be separated
well, and other separation methods should be sought out and applied
to overcome this limitation. In the functional analyses, it may be
important to clarify each of the proteins detailed role in hypoxic
regulation. Combination of proteomic studies and other experimental
techniques may be suitable to resolve this problem. Moreover,
database searching is the most common approach to identify the MS
detection data, but the results may be limited by the quality of
the protein database used. Proteins which are not annotated in the
database may not be identified and other identification methods,
such as spectral matching (86–89)
and de novo sequencing (18,90),
should be introduced to the identification process. Along with the
development of improved mass spectrometers and bioinformatic
algorithms, more and more valuable proteins may be identified by
MS-based proteomics in the future, which may offer a better
understanding of hypoxia pathways and their potential intervening
targets.
Acknowledgements
The present review was supported by grants from the
Major State Basic Research Development Program of China (no.
2013CB531503) and the National Key Research and Development Project
(no. 2016YFA0502203).
References
|
1
|
Carroll VA and Ashcroft M: Targeting the
molecular basis for tumour hypoxia. Expert Rev Mol Med. 7:1–16.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dewhirst MW, Cao Y and Moeller B: Cycling
hypoxia and free radicals regulate angiogenesis and radiotherapy
response. Nat Rev Cancer. 8:425–437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vaupel P and Mayer A: Hypoxia in cancer:
Significance and impact on clinical outcome. Cancer Metastasis Rev.
26:225–239. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bertout JA, Patel SA and Simon MC: The
impact of O2 availability on human cancer. Nat Rev
Cancer. 8:967–975. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Young SD, Marshall RS and Hill RP: Hypoxia
induces DNA overreplication and enhances metastatic potential of
murine tumor cells. Proc Natl Acad Sci USA. 85:9533–9537. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ruan K, Song G and Ouyang G: Role of
hypoxia in the hallmarks of human cancer. J Cell Biochem.
107:1053–1062. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vaupel P, Kelleher DK and Höckel M: Oxygen
status of malignant tumors: Pathogenesis of hypoxia and
significance for tumor therapy. Semin Oncol. 28:(Suppl 8). 29–35.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gray LH, Conger AD, Ebert M, Hornsey S and
Scott OC: The concentration of oxygen dissolved in tissues at the
time of irradiation as a factor in radiotherapy. Br J Radiol.
26:638–648. 1953. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moulder JE and Rockwell S: Tumor hypoxia:
Its impact on cancer therapy. Cancer Metastasis Rev. 5:313–341.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chaudary N and Hill RP: Hypoxia and
metastasis. Clin Cancer Res. 13:1947–1949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moyer MW: Targeting hypoxia brings breath
of fresh air to cancer therapy. Nat Med. 18:636–637. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wasinger VC, Cordwell SJ, Cerpa-Poljak A,
Yan JX, Gooley AA, Wilkins MR, Duncan MW, Harris R, Williams KL and
Humphery-Smith I: Progress with gene-product mapping of the
Mollicutes: Mycoplasma genitalium. Electrophoresis. 16:1090–1094.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen B, Zhang D, Wang X, Ma W, Deng S,
Zhang P, Zhu H, Xu N and Liang S: Proteomics progresses in
microbial physiology and clinical antimicrobial therapy. Eur J Clin
Microbiol Infect Dis. 36:403–413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ion A, Popa IM, Papagheorghe LM, Lisievici
C, Lupu M, Voiculescu V, Caruntu C and Boda D: Proteomic approaches
to biomarker discovery in cutaneous T-cell lymphoma. Dis Markers.
2016:96024722016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Tian W and Song J: Proteomics
applications in dental derived stem cells. J Cell Physiol.
232:1602–1610. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aebersold R and Mann M: Mass
spectrometry-based proteomics. Nature. 422:198–207. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cravatt BF, Simon GM and Yates JR III: The
biological impact of mass-spectrometry-based proteomics. Nature.
450:991–1000. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nesvizhskii AI, Vitek O and Aebersold R:
Analysis and validation of proteomic data generated by tandem mass
spectrometry. Nat Methods. 4:787–797. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shapiro AL, Viñuela E and Maizel JV Jr:
Molecular weight estimation of polypeptide chains by
electrophoresis in SDS-polyacrylamide gels. Biochem Biophys Res
Commun. 28:815–820. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shapiro HD, Miller KD and Harris AH:
Low-pH disc electrophoresis of spinal fluid; changes in multiple
sclerosis. Exp Mol Pathol. 7:362–365. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weber K and Osborn M: The reliability of
molecular weight determinations by dodecyl sulfate-polyacrylamide
gel electrophoresis. J Biol Chem. 244:4406–4412. 1969.PubMed/NCBI
|
|
22
|
O'Farrell PH: High resolution
two-dimensional electrophoresis of proteins. J Biol Chem.
250:4007–4021. 1975.PubMed/NCBI
|
|
23
|
Unlü M, Morgan ME and Minden JS:
Difference gel electrophoresis: A single gel method for detecting
changes in protein extracts. Electrophoresis. 18:2071–2077. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Alban A, David SO, Bjorkesten L, Andersson
C, Sloge E, Lewis S and Currie I: A novel experimental design for
comparative two-dimensional gel analysis: Two-dimensional
difference gel electrophoresis incorporating a pooled internal
standard. Proteomics. 3:36–44. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Strittmatter EF, Ferguson PL, Tang K and
Smith RD: Proteome analyses using accurate mass and elution time
peptide tags with capillary LC time-of-flight mass spectrometry. J
Am Soc Mass Spectrom. 14:980–991. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lundgren DH, Hwang SI, Wu L and Han DK:
Role of spectral counting in quantitative proteomics. Expert Rev
Proteomics. 7:39–53. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ong SE, Blagoev B, Kratchmarova I,
Kristensen DB, Steen H, Pandey A and Mann M: Stable isotope
labeling by amino acids in cell culture, SILAC, as a simple and
accurate approach to expression proteomics. Mol Cell Proteomics.
1:376–386. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schmidt A, Kellermann J and Lottspeich F:
A novel strategy for quantitative proteomics using isotope-coded
protein labels. Proteomics. 5:4–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wiese S, Reidegeld KA, Meyer HE and
Warscheid B: Protein labeling by iTRAQ: A new tool for quantitative
mass spectrometry in proteome research. Proteomics. 7:340–350.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thompson A, Schäfer J, Kuhn K, Kienle S,
Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK and Hamon
C: Tandem mass tags: A novel quantification strategy for
comparative analysis of complex protein mixtures by MS/MS. Anal
Chem. 75:1895–1904. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen Y, Shi G, Xia W, Kong C, Zhao S, Gaw
AF, Chen EY, Yang GP, Giaccia AJ, Le QT, et al: Identification of
hypoxia-regulated proteins in head and neck cancer by proteomic and
tissue array profiling. Cancer Res. 64:7302–7310. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Q, Van Antwerp D, Mercurio F, Lee KF
and Verma IM: Severe liver degeneration in mice lacking the IkappaB
kinase 2 gene. Science. 284:321–325. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Magagnin MG, Sergeant K, van den Beucken
T, Rouschop KM, Jutten B, Seigneuric R, Lambin P, Devreese B,
Koritzinsky M and Wouters BG: Proteomic analysis of gene expression
following hypoxia and reoxygenation reveals proteins involved in
the recovery from endoplasmic reticulum and oxidative stress.
Radiother Oncol. 83:340–345. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Woodman PG: p97, a protein coping with
multiple identities. J Cell Sci. 116:4283–4290. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Koritzinsky M, Magagnin MG, van den
Beucken T, Seigneuric R, Savelkouls K, Dostie J, Pyronnet S,
Kaufman RJ, Weppler SA, Voncken JW, et al: Gene expression during
acute and prolonged hypoxia is regulated by distinct mechanisms of
translational control. EMBO J. 25:1114–1125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Koritzinsky M, Seigneuric R, Magagnin MG,
van den Beucken T, Lambin P and Wouters BG: The hypoxic proteome is
influenced by gene-specific changes in mRNA translation. Radiother
Oncol. 76:177–186. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lü X, de la Peña L, Barker C, Camphausen K
and Tofilon PJ: Radiation-induced changes in gene expression
involve recruitment of existing messenger RNAs to and away from
polysomes. Cancer Res. 66:1052–1061. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nordsmark M, Loncaster J, Aquino-Parsons
C, Chou SC, Gebski V, West C, Lindegaard JC, Havsteen H, Davidson
SE, Hunter R, et al: The prognostic value of pimonidazole and
tumour pO2 in human cervix carcinomas after
radiation therapy: A prospective international multi-center study.
Radiother Oncol. 80:123–131. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Magagnin MG, van den Beucken T, Sergeant
K, Lambin P, Koritzinsky M, Devreese B and Wouters BG: The mTOR
target 4E-BP1 contributes to differential protein expression during
normoxia and hypoxia through changes in mRNA translation
efficiency. Proteomics. 8:1019–1028. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Semenza GL, Jiang BH, Leung SW, Passantino
R, Concordet JP, Maire P and Giallongo A: Hypoxia response elements
in the aldolase A, enolase 1, and lactate dehydrogenase A gene
promoters contain essential binding sites for hypoxia-inducible
factor 1. J Biol Chem. 271:32529–32537. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Buono RJ and Lang RK: Hypoxic repression
of lactate dehydrogenase-B in retina. Exp Eye Res. 69:685–693.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Choi KJ, Piao YJ, Lim MJ, Kim JH, Ha J,
Choe W and Kim SS: Overexpressed cyclophilin A in cancer cells
renders resistance to hypoxia- and cisplatin-induced cell death.
Cancer Res. 67:3654–3662. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dundas SR, Lawrie LC, Rooney PH and Murray
GI: Mortalin is over-expressed by colorectal adenocarcinomas and
correlates with poor survival. J Pathol. 205:74–81. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cui Y, Zhang D, Jia Q, Li T, Zhang W and
Han J: Proteomic and tissue array profiling identifies elevated
hypoxia-regulated proteins in pancreatic ductal adenocarcinoma.
Cancer Invest. 27:747–755. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Vaupel P: The role of hypoxia-induced
factors in tumor progression. Oncologist. 9:(Suppl 5). S10–S17.
2004. View Article : Google Scholar
|
|
46
|
Hu R, Jin H, Zhou S, Yang P and Li X:
Proteomic analysis of hypoxia-induced responses in the
syncytialization of human placental cell line BeWo. Placenta.
28:399–407. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Furuta E, Pai SK, Zhan R, Bandyopadhyay S,
Watabe M, Mo YY, Hirota S, Hosobe S, Tsukada T, Miura K, et al:
Fatty acid synthase gene is up-regulated by hypoxia via activation
of Akt and sterol regulatory element binding protein-1. Cancer Res.
68:1003–1011. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee AS: Mammalian stress response:
Induction of the glucose-regulated protein family. Curr Opin Cell
Biol. 4:267–273. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fajardo I, Svensson L, Bucht A and Pejler
G: Increased levels of hypoxia-sensitive proteins in allergic
airway inflammation. Am J Respir Crit Care Med. 170:477–484. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Larsen M, Tazzyman S, Lund EL, Junker N,
Lewis CE, Kristjansen PE and Murdoch C: Hypoxia-induced secretion
of macrophage migration-inhibitory factor from MCF-7 breast cancer
cells is regulated in a hypoxia-inducible factor-independent
manner. Cancer Lett. 265:239–249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liao SH, Zhao XY, Han YH, Zhang J, Wang
LS, Xia L, Zhao KW, Zheng Y, Guo M and Chen GQ: Proteomics-based
identification of two novel direct targets of hypoxia-inducible
factor-1 and their potential roles in migration/invasion of cancer
cells. Proteomics. 9:3901–3912. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pellieux C, Desgeorges A, Pigeon CH,
Chambaz C, Yin H, Hayoz D and Silacci P: Cap G, a gelsolin family
protein modulating protective effects of unidirectional shear
stress. J Biol Chem. 278:29136–29144. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Garrett SC, Varney KM, Weber DJ and
Bresnick AR: S100A4, a mediator of metastasis. J Biol Chem.
281:677–680. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tarabykina S, Griffiths TR, Tulchinsky E,
Mellon JK, Bronstein IB and Kriajevska M: Metastasis-associated
protein S100A4: Spotlight on its role in cell migration. Curr
Cancer Drug Targets. 7:217–228. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bartkowiak K, Effenberger KE, Harder S,
Andreas A, Buck F, Peter-Katalinic J, Pantel K and Brandt BH:
Discovery of a novel unfolded protein response phenotype of cancer
stem/progenitor cells from the bone marrow of breast cancer
patients. J Proteome Res. 9:3158–3168. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Grandjean M, Sermeus A, Branders S,
Defresne F, Dieu M, Dupont P, Raes M, De Ridder M and Feron O:
Hypoxia integration in the serological proteome analysis unmasks
tumor antigens and fosters the identification of anti-phospho-eEF2
antibodies as potential cancer biomarkers. PLoS One. 8:e765082013.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yoon JH, Kim J, Kim KL, Kim DH, Jung SJ,
Lee H, Ghim J, Kim D, Park JB, Ryu SH, et al: Proteomic analysis of
hypoxia-induced U373MG glioma secretome reveals novel
hypoxia-dependent migration factors. Proteomics. 14:1494–1502.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chang AC, Janosi J, Hulsbeek M, de Jong D,
Jeffrey KJ, Noble JR and Reddel RR: A novel human cDNA highly
homologous to the fish hormone stanniocalcin. Mol Cell Endocrinol.
112:241–247. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Varghese R, Wong CK, Deol H, Wagner GF and
DiMattia GE: Comparative analysis of mammalian stanniocalcin genes.
Endocrinology. 139:4714–4725. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tamura S, Oshima T, Yoshihara K, Kanazawa
A, Yamada T, Inagaki D, Sato T, Yamamoto N, Shiozawa M, Morinaga S,
et al: Clinical significance of STC1 gene expression in
patients with colorectal cancer. Anticancer Res. 31:325–329.
2011.PubMed/NCBI
|
|
61
|
Nakagawa T, Martinez SR, Goto Y, Koyanagi
K, Kitago M, Shingai T, Elashoff DA, Ye X, Singer FR, Giuliano AE,
et al: Detection of circulating tumor cells in early-stage breast
cancer metastasis to axillary lymph nodes. Clin Cancer Res.
13:4105–4110. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cheng J, Gao F, Chen X, Wu J, Xing C, Lv
Z, Xu W, Xie Q, Wu L, Ye S, et al: Prohibitin-2 promotes
hepatocellular carcinoma malignancy progression in hypoxia based on
a label-free quantitative proteomics strategy. Mol Carcinog.
53:820–832. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bogenhagen DF, Wang Y, Shen EL and
Kobayashi R: Protein components of mitochondrial DNA nucleoids in
higher eukaryotes. Mol Cell Proteomics. 2:1205–1216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
McClung JK, Jupe ER, Liu XT and Dell'Orco
RT: Prohibitin: Potential role in senescence, development, and
tumor suppression. Exp Gerontol. 30:99–124. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Nijtmans LG, de Jong L, Sanz Artal M,
Coates PJ, Berden JA, Back JW, Muijsers AO, van der Spek H and
Grivell LA: Prohibitins act as a membrane-bound chaperone for the
stabilization of mitochondrial proteins. EMBO J. 19:2444–2451.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Osman C, Merkwirth C and Langer T:
Prohibitins and the functional compartmentalization of
mitochondrial membranes. J Cell Sci. 122:3823–3830. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Valli A, Rodriguez M, Moutsianas L,
Fischer R, Fedele V, Huang HL, Van Stiphout R, Jones D, Mccarthy M,
Vinaxia M, et al: Hypoxia induces a lipogenic cancer cell phenotype
via HIF1α-dependent and -independent pathways. Oncotarget.
6:1920–1941. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Delcourt N, Quevedo C, Nonne C, Fons P,
O'Brien D, Loyaux D, Diez M, Autelitano F, Guillemot JC, Ferrara P,
et al: Targeted identification of sialoglycoproteins in hypoxic
endothelial cells and validation in zebrafish reveal roles for
proteins in angiogenesis. J Biol Chem. 290:3405–3417. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nagao K and Oka K: HIF-2 directly
activates CD82 gene expression in endothelial cells. Biochem
Biophys Res Commun. 407:260–265. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lee P, Goishi K, Davidson AJ, Mannix R,
Zon L and Klagsbrun M: Neuropilin-1 is required for vascular
development and is a mediator of VEGF-dependent angiogenesis in
zebrafish. Proc Natl Acad Sci USA. 99:10470–10475. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mura M, Swain RK, Zhuang X, Vorschmitt H,
Reynolds G, Durant S, Beesley JF, Herbert JM, Sheldon H, Andre M,
et al: Identification and angiogenic role of the novel tumor
endothelial marker CLEC14A. Oncogene. 31:293–305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Stockwin LH, Blonder J, Bumke MA, Lucas
DA, Chan KC, Conrads TP, Issaq HJ, Veenstra TD, Newton DL and Rybak
SM: Proteomic analysis of plasma membrane from hypoxia-adapted
malignant melanoma. J Proteome Res. 5:2996–3007. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yang W, Thompson JW, Wang Z, Wang L, Sheng
H, Foster MW, Moseley MA and Paschen W: Analysis of
oxygen/glucose-deprivation-induced changes in SUMO3 conjugation
using SILAC-based quantitative proteomics. J Proteome Res.
11:1108–1117. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Djidja MC, Chang J, Hadjiprocopis A,
Schmich F, Sinclair J, Mršnik M, Schoof EM, Barker HE, Linding R,
Jørgensen C, et al: Identification of hypoxia-regulated proteins
using MALDI-mass spectrometry imaging combined with quantitative
proteomics. J Proteome Res. 13:2297–2313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Malec V, Coulson JM, Urbé S and Clague MJ:
Combined analyses of the VHL and hypoxia signaling axes in an
isogenic pairing of renal clear cell carcinoma cells. J Proteome
Res. 14:5263–5272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Chan DA, Sutphin PD, Nguyen P, Turcotte S,
Lai EW, Banh A, Reynolds GE, Chi JT, Wu J, Solow-Cordero DE, et al:
Targeting GLUT1 and the Warburg effect in renal cell carcinoma by
chemical synthetic lethality. Sci Transl Med. 3:94ra702011.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yamasaki T, Seki N, Yoshino H, Itesako T,
Yamada Y, Tatarano S, Hidaka H, Yonezawa T, Nakagawa M and Enokida
H: Tumor-suppressive microRNA-1291 directly regulates
glucose transporter 1 in renal cell carcinoma. Cancer Sci.
104:1411–1419. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hosoya N, Sakumoto M, Nakamura Y, Narisawa
T, Bilim V, Motoyama T, Tomita Y and Kondo T: Proteomics identified
nuclear N-myc downstream-regulated gene 1 as a prognostic tissue
biomarker candidate in renal cell carcinoma. Biochim Biophys Acta.
1834:2630–2639. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ren Y, Hao P, Dutta B, Cheow ES, Sim KH,
Gan CS, Lim SK and Sze SK: Hypoxia modulates A431 cellular pathways
association to tumor radioresistance and enhanced migration
revealed by comprehensive proteomic and functional studies. Mol
Cell Proteomics. 12:485–498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lara PC, Lloret M, Clavo B, Apolinario RM,
Bordón E, Rey A, Falcón O, Alonso AR and Belka C: Hypoxia
downregulates Ku70/80 expression in cervical carcinoma tumors.
Radiother Oncol. 89:222–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Park JE, Tan HS, Datta A, Lai RC, Zhang H,
Meng W, Lim SK and Sze SK: Hypoxic tumor cell modulates its
microenvironment to enhance angiogenic and metastatic potential by
secretion of proteins and exosomes. Mol Cell Proteomics.
9:1085–1099. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ren Y, Hao P, Law SK and Sze SK:
Hypoxia-induced changes to integrin α 3 glycosylation facilitate
invasion in epidermoid carcinoma cell line A431. Mol Cell
Proteomics. 13:3126–3137. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Dutta B, Yan R, Lim SK, Tam JP and Sze SK:
Quantitative profiling of chromatome dynamics reveals a novel role
for HP1BP3 in hypoxia-induced oncogenesis. Mol Cell Proteomics.
13:3236–3249. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yanagisawa K, Konishi H, Arima C, Tomida
S, Takeuchi T, Shimada Y, Yatabe Y, Mitsudomi T, Osada H and
Takahashi T: Novel metastasis-related gene CIM functions in the
regulation of multiple cellular stress-response pathways. Cancer
Res. 70:9949–9958. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
McMahon KM, Volpato M, Chi HY, Musiwaro P,
Poterlowicz K, Peng Y, Scally AJ, Patterson LH, Phillips RM and
Sutton CW: Characterization of changes in the proteome in different
regions of 3D multicell tumor spheroids. J Proteome Res.
11:2863–2875. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Yates JR III, Morgan SF, Gatlin CL,
Griffin PR and Eng JK: Method to compare collision-induced
dissociation spectra of peptides: Potential for library searching
and subtractive analysis. Anal Chem. 70:3557–3565. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Craig R, Cortens JC, Fenyo D and Beavis
RC: Using annotated peptide mass spectrum libraries for protein
identification. J Proteome Res. 5:1843–1849. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Frewen BE, Merrihew GE, Wu CC, Noble WS
and MacCoss MJ: Analysis of peptide MS/MS spectra from large-scale
proteomics experiments using spectrum libraries. Anal Chem.
78:5678–5684. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Lam H, Deutsch EW, Eddes JS, Eng JK, King
N, Stein SE and Aebersold R: Development and validation of a
spectral library searching method for peptide identification from
MS/MS. Proteomics. 7:655–667. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Nesvizhskii AI, Roos FF, Grossmann J,
Vogelzang M, Eddes JS, Gruissem W, Baginsky S and Aebersold R:
Dynamic spectrum quality assessment and iterative computational
analysis of shotgun proteomic data: Toward more efficient
identification of post-translational modifications, sequence
polymorphisms, and novel peptides. Mol Cell Proteomics. 5:652–670.
2006. View Article : Google Scholar : PubMed/NCBI
|