Introduction
Diffuse large B-cell lymphoma (DLBCL) is a type of
non-Hodgkin lymphoma (NHL), which is most frequently diagnosed in
adults and comprises 35–40% of all NHL cases in Western countries
(1). Germinal center B-cell
(GCB)-like DLBCL is a subtype of DLBCL and is postulated to arise
from GC centroblasts, which is characterized by high expression
level of BCL6 and somatic hypermutations of immunoglobulin
genes (2,3). The most prevalent chromosomal
translocation found in GCB-DLBCL is t(14;18)(q32;q21) which can be
detected in 30–40% of the cases (4). Moreover, 8q24 rearrangement involving
MYC (5) and 10q23 deletion
involving PTEN (6) have also
been detected in GCB-DLBCL. Enhancer of zeste homolog 2
(EZH2), an epigenetic modifier, exhibits gain-of-function
mutations in 6–14% of DLBCL cases, and these mutations are detected
almost exclusively in the GCB subtype (7,8).
However, the presence of these genetic aberrations does not fully
explain the complex pathogenesis of GCB-DLBCL, for the reason that
this disease may also partially result from epigenetic factors such
as cytosine modifications, histone modifications, and the influence
of non-coding RNA molecules.
Non-coding RNAs (ncRNAs) are thought to be
critically involved in both cellular physiological processes and
cancer pathogenesis. These ncRNAs exist in two forms: short ncRNAs
that comprise 18–200 nucleotides and long ncRNAs that comprise
>200 nucleotides (9).
Additionally, a variety of microRNAs (miRs) such as miR-155 and
miR-17–92 have been shown to be aberrantly expressed in GCB-DLBCL
(10). Accumulating data have
suggested the critical roles of long non-coding RNAs (lncRNAs) in
immune responses, cell differentiation, tumorigenesis and genomic
imprinting (11). Moreover,
aberrantly expressed lncRNAs have also been shown to be associated
with certain types of cancers including pancreatic cancer,
glioblastoma and hepatocellular carcinoma (11–13).
The functions and genome-wide expression patterns of
lncRNAs in GCB-DLBCL have remained largely unclear. Therefore, we
performed microarray analysis to investigate the expression
profiles of lncRNAs in GCB-DLBCL cells. Our results showed that
thousands of differentially expressed lncRNAs were present in
GCB-DLBCL cells compared with benign B cells. Next, 8 lncRNAs were
selected to validate the microarray results by qRT-PCR.
Furthermore, the possible functions and mechanisms of these lncRNAs
were predicted by Gene Ontology, pathway and network analyses.
Conclusively, our findings may offer further insight into the
occurrence and development of GCB-DLBCL.
Materials and methods
Sample preparation and RNA
extraction
The methods used in the present study conformed with
ethical principles listed in the Helsinki Declaration, and the
study protocol was reviewed and approved by the China Medical
University Ethics Committee. Ten biopsy specimens of GCB-DLBCL
tissue and 10 specimens of reactive lymph node (RLN) tissue were
included in the present study, in addition to the cell lines
described below. The biopsy specimens were provided by the
Shengjing Hospital of China Medical University, Department of
Pathology. Criteria described in the 2008 World Health Organization
(WHO) classification system were used to confirm all diagnoses of
GCB-DLBCL. Human GCB-DLBCL cell lines OCI-ly1 and OCI-ly19 were
purchased from the Shanghai Institutes for Biological Sciences Cell
Resource Center and cultured in Dulbecco's modified Eagles medium
(DMEM)-high glucose which contained 10% fetal bovine serum (FBS;
Gibco, Carlsbad, CA, USA). Healthy donor blood was provided by the
Regional Blood Donor Center, and CD19 microbeads (Miltenyi Biotec,
Bergisch Gladbach, Germany) were used to isolate CD19-positive B
cells from the buffy coats of those donor samples. All cells were
incubated in a 37°C incubator with a humidified atmosphere of 5%
CO2.
The total RNA of each sample was isolated using
TRIzol reagent (Invitrogen, Carlsbad, CA, USA); after which, the
amounts of extracted RNA were quantified by a NanoDrop ND-1000
spectrophotometer. The structural integrity of isolated RNA was
evaluated by agarose gel electrophoresis under denaturing
conditions, and the RNA purity was assessed by the ratio of
absorbance at 260 and 280 nm.
RNA microarray
Arraystar Human LncRNA Microarray V3.0 was used to
profile the lncRNAs in our specimens, and was able to detect
~30,586 lncRNAs and 26,109 coding transcripts. Information provided
in various transcriptome databases (RefSeq, GENCODE and UCSC
Knowngenes) and previously published studies was used to help
construct the lncRNAs. The individual transcripts were accurately
identified by highly specific exon or splice junction probes. To
ensure the quality control of hybridization, the array also
included negative probes and positive probes for housekeeping
genes.
RNA labeling and array
hybridization
A slightly modified version of the Agilent One-Color
Microarray-Based Gene Expression Analysis protocol (Agilent
Technologies, Inc., Santa Clara, CA, USA) was used for sample
labeling and array hybridization. In brief, an mRNA-ONLY™
Eukaryotic mRNA Isolation kit (Epicentre Biotechnologies, Madison,
WI, USA) was used to remove rRNA from total RNA. Next, an Arraystar
Flash RNA Labeling kit (Arraystar, Rockville, MD, USA) was used to
amplify each purified sample and then transcribe it into
fluorescent cRNA containing the full length transcript without 3′
bias using a random priming method. The fluorescence-labeled cRNAs
were purified using an RNeasy Mini kit (Qiagen, Valencia, CA, USA).
Spectrophotometric methods were used to measure the concentration
and specific activity of the labeled cRNAs (pmol Cy3/µg cRNA). One
microgram of the labeled cRNAs was treated with 5 µl of 10X
blocking agent and 1 µl of 25X fragmentation buffer; after which,
the mixture was heated for 30 min at 60°C. After heating, the
mixture was diluted with 25 µl of 2X GE hybridization buffer.
Subsequently, 50 µl of hybridization solution was dispensed into
the gasket slide and assembled to the lncRNA expression microarray
slide. The combined slides were then incubated in an Agilent
hybridization oven set at 65°C for 17 h. Following incubation, the
hybridized arrays were washed, fixed and scanned with an Agilent
DNA Microarray Scanner (part no. G2505C).
Analysis of microarray data
All the array images were analyzed using Agilent
Feature Extraction software (version 11.0.1.1). GeneSpring GX v12.1
software (Agilent Technologies) was used to normalize quartile
values and perform the required data processing. After the raw data
had been normalized, lncRNAs and mRNAs that had flags indicating
either ‘Present’ or ‘Marginal’ in ≥3 of 9 samples were selected for
further evaluation. LncRNAs and mRNAs that showed significant
differential expression patterns in lymphoma cells vs. normal cells
were identified by P-value/FDR filtering and fold-change filtering.
Homemade scripts were used for purposes of hierarchical clustering
and combined analyses.
Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway analysis
The Gene Ontology (GO) consortium (http://www.geneontology.org) has developed a
comprehensive unified vocabulary for use when describing genes and
gene products of an organism. This ontology classifies functions
along three aspects: biological process, cellular component and
molecular function. We used Fisher's exact test to determine
whether any overlap that existed among items on the differentially
expressed (DE) list and GO annotation list was larger than could be
expected to occur by chance. The statistical significance of the GO
term enrichment in the DE genes was expressed as the P-value, and
the FDR signifies the false discovery rate. A lower P-value
signifies a more significant GO term, with P-value ≤0.05 being
recommended. Pathway analysis was performed to map specific genes
to various KEGG pathways (http://www.genome.jp/kegg/). The P-value (EASE-score,
Fisher or hypergeometric P-values) was used to indicate the
significance of any correlation between a pathway and certain
conditions. A lower P-value indicates a more significant
correlation, with P-value ≤0.05 being recommended.
Quantitative reverse transcription
polymerase chain reaction (qRT-PCR)
Several lncRNAs from the microarray data analysis
were further validated by qRT-PCR. SuperScript™ III Reverse
Transcriptase (Invitrogen) was used for reverse transcription of
total RNA. Quantitative RT-PCR was performed using SYBR-Green kit
(Invitrogen) in a Rotor-Gene 3000 Real-time PCR Detection System
(Corbett Research, Brisbane, Australia). The specific primers used
to transcribe each gene are shown in Table I.
 | Table I.Oligonucleotide sequences of the
qRT-PCR primers. |
Table I.
Oligonucleotide sequences of the
qRT-PCR primers.
| Gene name | Bidirectional
primer sequences |
|---|
| β-actin | F:
5′-GTGGCCGAGGACTTTGATTG-3′ |
|
| R:
5′-CCTGTAACAACGCATCTCATATT-3′ |
|
ENST00000424690 | F:
5′-AGCCAACTGGGATAAAGAAGAG-3′ |
|
| R:
5′-TAGAGGAAGGCAACTGTCACTCT-3′ |
|
ENST00000455011 | F:
5′-AAAATGCGTGCCCTCTTGT-3′ |
|
| R:
5′-GGGCTCCATCATTCATCCTT-3′ |
|
ENST00000451368 | F:
5′-AAGCCTGAGTAACAGAGGAGAAC-3′ |
|
| R:
5′-CAACTGCACTCCAAATGGCT-3′ |
|
ENST00000425358 | F:
5′-CCAGAAACCAGCCATAGTCC-3′ |
|
| R:
5′-CCTTCCTCCGCTAAATCTCA-3′ |
|
ENST00000558952 | F:
5′-TTAGCCATACTCAGCACCCTT-3′ |
|
| R:
5′-CATGCACGTCAACCACAAAC-3′ |
| NR_026892 | F:
5′-CTTCCCTAACACCGCAGTATC-3′ |
|
| R:
5′-TTCCTAGACAGTCAGGCTCCAG-3′ |
|
ENST00000464929 | F:
5′-CCAAGATGGTTTCCCTAGAAG-3′ |
|
| R:
5′-GTCCAATATCAAGGTGTCAGCA-3′ |
|
ENST00000475089 | F:
5′-TCGGTGTCACATGCTGCTTC-3′ |
|
| R:
5′-GAGAACCCGATCCCTCCCTC-3′ |
The relative fold-change normalized to β-actin was
calculated using the 2−ΔΔCt method. Differences between
the lncRNAs expressed in groups of GCB-DLBCL cells vs. normal cells
were evaluated with the paired t-test. P-value <0.05 was
regarded as statistically significant.
Establishment of the co-expression
network
The correlations identified between differentially
expressed mRNAs and lncRNAs were used to construct the lncRNA-mRNA
co-expression network comprising 5 validated lncRNAs and their
co-expressed coding genes. The following steps were taken to
establish this network: i) we first pre-processed the data using
the median gene expression value of all transcripts expressed from
the same coding gene, without giving special treatment to the
lncRNA expression value. ii) Next, the data were screened for
differentially expressed mRNAs and lncRNAs, and then these data
were deleted from the dataset. iii) We calculated Pearson
correlation coefficient (PCC) and used the R-value to calculate the
correlation coefficient of the PCC between the lncRNAs and mRNAs
(only lncRNA-mRNA PCC, not including lncRNA-lncRNA and mRNA-mRNA
PCC). iv) Lastly, we defined PCC ≥0.99 as significant and then used
Cytoscape v2.8.1 software to plot the co-expression network.
Statistical analysis
All data were analyzed using IBM SPSS Statistics for
Windows, version 21.0 (IBM, Armonk, NY, USA). Student's paired
t-test was used to compare the parameters in the two groups.
P-value <0.05 was regarded as statistically significant.
Results
Differentially expressed lncRNAs
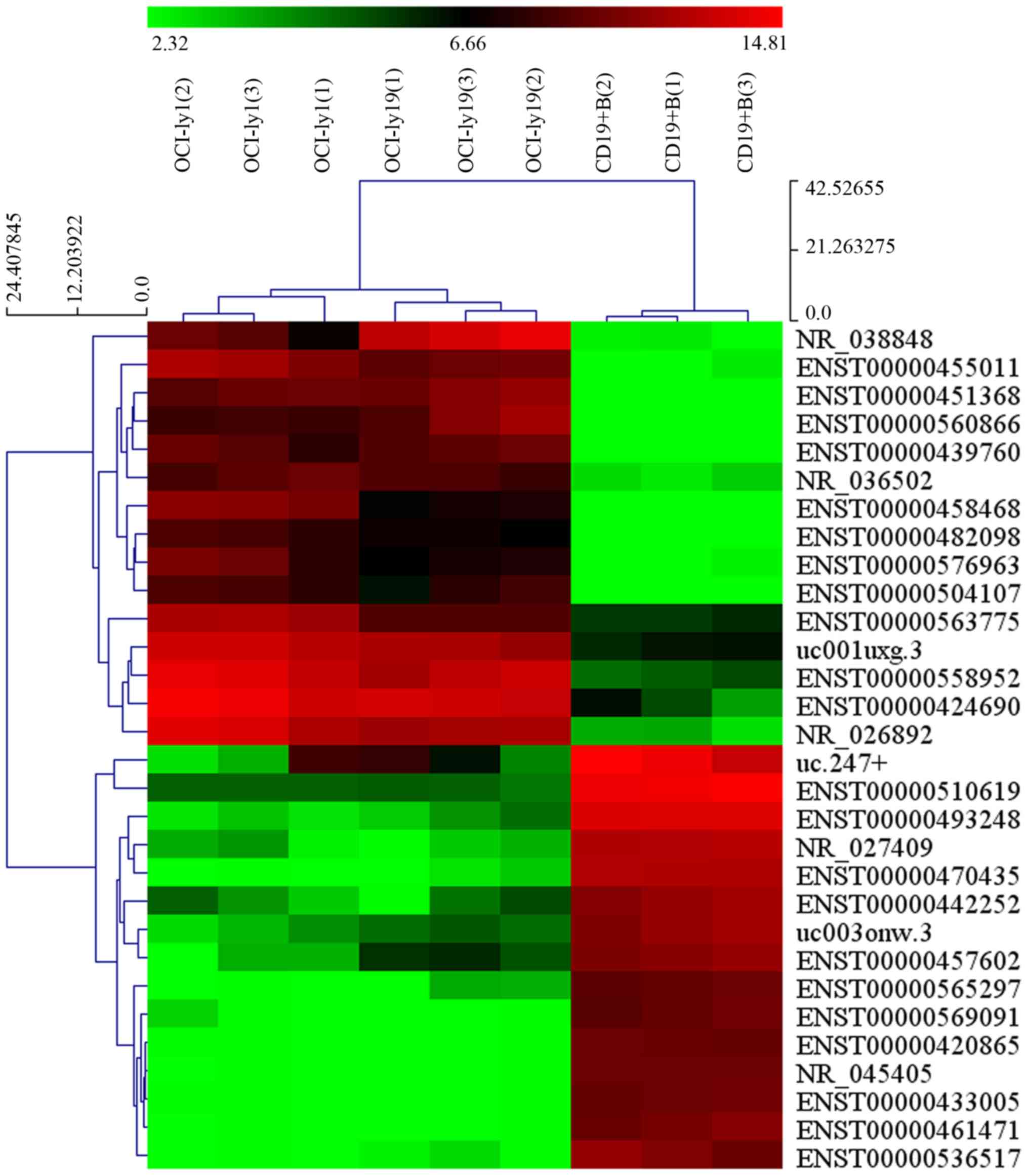
Our microarray results revealed that thousands of
differentially expressed lncRNAs were present in OCI-ly1 and
OCI-ly19 cells compared with benign B cells. Among them, 1,648
lncRNAs displayed significant upregulation, and 2,671 lncRNAs
displayed significant downregulation in the two lymphoma cell lines
(fold-change ≥2.0; P<0.05). NR_026892 and ENST00000464929 were
among the most upregulated and downregulated lncRNAs, respectively.
The heat map showing the results of a two-way hierarchical
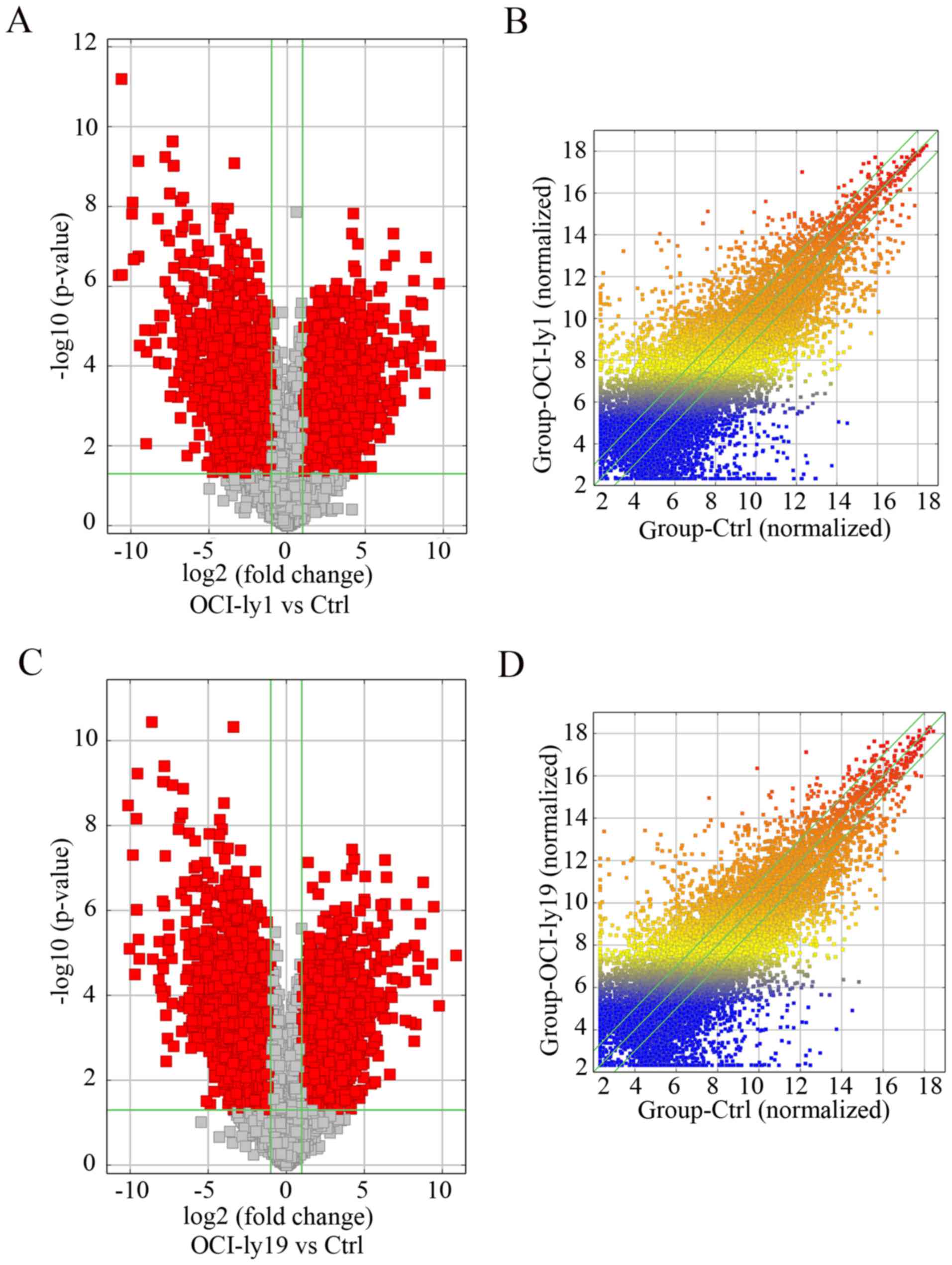
clustering of samples and lncRNAs is presented in Fig. 1. Additionally, the volcano plots
(Fig. 2A and C) and scatter plots
(Fig. 2B and D) display the
aberrantly expressed lncRNAs between lymphoma cells and normal
cells.
For further analysis, the lncRNAs were classified
into three different groups: antisense, enhancer and lincRNAs,
respectively. We found that 612 antisense lncRNAs, 333 enhancer
lncRNAs and 581 lincRNAs were deregulated in the OCI-ly1 cells, and
694 antisense lncRNAs, 349 enhancer lncRNAs and 646 lincRNAs were
deregulated in the OCI-ly19 cells (fold-change ≥2.0;
P<0.05).
Differentially expressed mRNAs
The differentially expressed mRNAs in the OCI-ly1
and OCI-ly19 cells compared with normal B lymphocytes were
identified using microarray technology. Our mRNA expression
profiling data indicated that 3,691 mRNAs displayed significant
upregulation, and 2,974 mRNAs displayed significant downregulation
in the two lymphoma cell lines (fold-change ≥2.0; P<0.05).
NM_001071 was one of the most upregulated mRNAs, while NM_033512
was one of the most downregulated mRNAs.
GO and KEGG pathway analysis
Our GO analysis showed that the most enriched GO
terms targeted by upregulated transcripts referred to
single-organism process (ontology: biological process), cell part
and cell (ontology: cellular component) and catalytic activity
(ontology: molecular function). Similarly, the most enriched GO
terms targeted by downregulated transcripts referred to
single-organism process (ontology: biological process), cell part
and cell (ontology: cellular component) and binding (ontology:
molecular function) (Fig. 3).
Our KEGG pathway analysis indicated that the
upregulated transcripts corresponded to 64 pathways. The
‘proteasome pathway’ (Pathway ID: hsa03050) which included 32
upregulated genes in our profiles was the highest enriched pathway
associated with tumors (Fig. 4A).
However, our analysis also revealed that the downregulated
transcripts corresponded to 62 pathways. The ‘MAPK signaling
pathway’ (Pathway ID: hsa04010) which included 69 downregulated
genes in our profiles showed the greatest enrichment among those
pathways associated with tumors (Fig.
4B).
Validation of microarray results by
qRT-PCR
The microarray results of 8 selected lncRNAs were
further validated by qRT-PCR. The selected lncRNAs included 6
upregulated and 2 downregulated lncRNAs (Table II). On the one hand, we selected 2
upregulated lncRNAs (ENST00000455011 and ENST00000451368) and 2
downregulated lncRNAs (ENST00000464929 and ENST00000475089) due to
their significant fold-change in our expression profiles. On the
other hand, we also selected 4 upregulated lncRNAs
(ENST00000558952, ENST00000425358, NR_026892 and ENST00000424690)
from our profiles since all of them were reported to either act as
carcinogenic factors or be associated with poor clinical outcomes
in other types of tumors. Data analysis indicated a statistically
significant difference. The qRT-PCR results showed that
ENST00000424690, ENST00000425358 and NR_026892 were upregulated,
while ENST00000464929 and ENST00000475089 were downregulated in
OCI-ly1 and OCI-ly19 cells compared with normal B lymphocytes
(fold-change ≥2.0; P<0.05) (Fig. 5A
and B).
| Table II.Basic information of the selected 8
lncRNAs for qRT-PCR confirmation. |
Table II.
Basic information of the selected 8
lncRNAs for qRT-PCR confirmation.
|
|
| OCI-ly1 cells | OCI-ly19 cells |
|---|
|
|
|
|
|
|---|
| Seqname | Regulation | P-value | Fold-change | P-value | Fold-change |
|---|
|
ENST00000424690 | Up | 0.000479464 | 446.7613383 | 0.000428945 | 251.1963515 |
|
ENST00000455011 | Up | 3.20045E-05 | 582.2601741 | 7.7906E-06 | 196.3856176 |
|
ENST00000451368 | Up | 5.46548E-06 | 190.1528344 | 2.62789E-05 | 351.5618304 |
|
ENST00000425358 | Up | 2.05718E-05 | 53.1231945 | 3.4208E-06 | 44.0691755 |
|
ENST00000558952 | Up | 2.29737E-05 | 370.505128 | 4.90638E-05 | 176.043018 |
| NR_026892 | Up | 9.4681E-05 | 874.6597124 | 1.17842E-05 | 345.194388 |
|
ENST00000464929 | Down | 6.4E-12 | 1563.86804 | 7.95406E-06 | 1055.412968 |
|
ENST00000475089 | Down | 7.8724E-09 | 960.5307121 | 4.91065E-08 | 914.0689387 |
Additionally, qRT-PCR was used to detect the
expression levels of these 5 lncRNAs (ENST00000424690,
ENST00000425358, NR_026892, ENST00000464929 and ENST00000475089) in
10 pairs of clinical samples including 10 GCB-DLBCL samples and 10
RLN samples. Our results showed that ENST00000424690,
ENST00000425358 and NR_026892 were upregulated, while
ENST00000464929 and ENST00000475089 were downregulated in the
GCB-DLBCL samples compared with the RLN samples (fold-change ≥2.0;
P<0.05) (Fig. 5C). Thus, the
expression trend of these 5 lncRNAs in clinical samples was
essentially the same as indicated by the microarray data.
Establishment of the lncRNA-mRNA
co-expression network
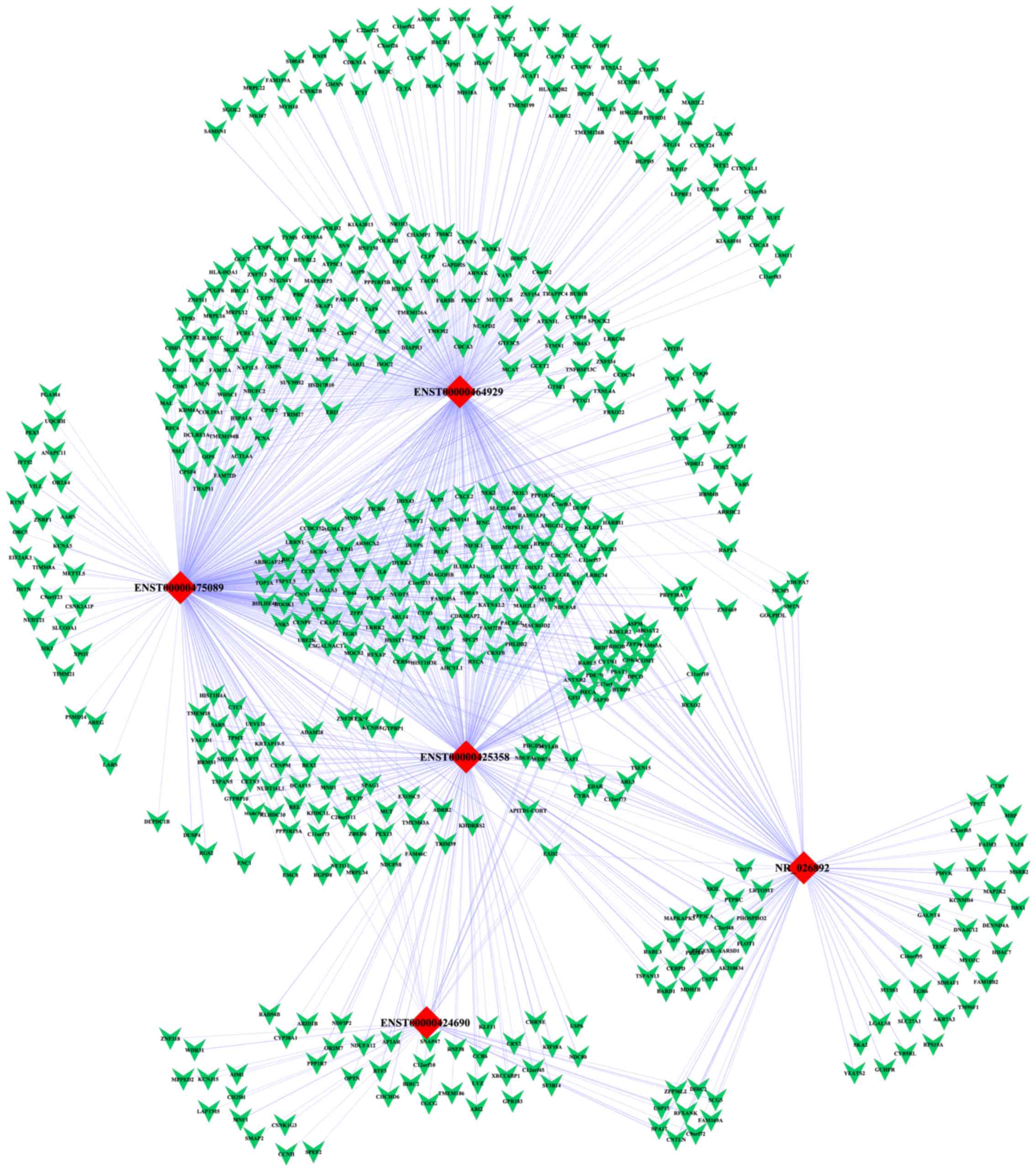
We constructed a lncRNA-mRNA co-expression network
consisting of 5 deregulated lncRNAs and their co-expressed coding
genes. As we mentioned above, 6 upregulated lncRNAs and 2
downregulated lncRNAs were chosen for further confirmation of
microarray results using qRT-PCR. However, 3 of them were not shown
to be differentially expressed in GCB-DLBCL cells according to the
qRT-PCR results, for the reason that false-positive results did
exist in the high throughput detection by microarray. Therefore, we
only selected 5 validated lncRNAs to construct the co-expression
network. The mRNAs and lncRNAs with PCC ≥0.99 were selected to
establish the network using Cytoscape software. The network
comprised 522 nodes and 1,005 connections between 517 mRNAs and 5
lncRNAs. Among the connections, 434 and 571 pairs presented as
positive and negative, respectively (Fig. 6). Our co-expression network
suggested that inter-regulation of lncRNAs and coding genes may
contribute to GCB-DLBCL pathogenesis.
Discussion
DLBCL is the most common type of NHL in adults and
is characterized by a wide variety of genetic aberrations including
BCL2 translocation (4) and somatic
hypermutations of immunoglobulin genes (2,3).
Although the molecular mechanisms underlying the occurrence and
development of GCB-DLBCL have been extensively investigated over
the past decades, its pathogenesis has not been fully elucidated.
To date, increasing evidence indicates that aberrantly expressed
lncRNAs are associated with tumorigenesis and chemotherapy
resistance in various cancers, such as HOTAIR in breast cancer
(14), CCAT1 in colorectal cancer
(15), HEIH in hepatocellular
carcinoma (12) and MALAT1 in DLBCL
(16). Moreover, the biological
functions of several deregulated lncRNAs in lymphoid malignancies
have been well documented, including LUNAR1 (17), MINCR (18) and FAS-AS1 (19). Recently, it has been reported that
immune-related lncRNA biomarkers are associated with both the
molecular subtype of DLBCL and the prognosis of DLBCL patients
(20).
In the present study, we applied microarray
technology to investigate the expression patterns of lncRNAs in two
types of GCB-DLBCL cell lines and identified 1,648 upregulated
lncRNAs and 2,671 downregulated lncRNAs. We then performed qRT-PCR
to validate the expression levels of 5 selected lncRNAs
(ENST00000424690, ENST00000425358, NR_026892, ENST00000464929 and
ENST00000475089) in the same series of samples, as well as in 10
pairs of clinical samples. The qRT-PCR results were essentially
consistent with those obtained from the microarray analysis.
Subsequently, our KEGG pathway analysis showed that totals of 64
and 62 biological pathways corresponded to the upregulated and
downregulated transcripts, respectively.
We focused on the ubiquitin-proteasome and p53
signaling pathways due to the fact that the ubiquitin-proteasome
system exerts critical roles particularly in hematological
malignancies and inactivation of p53 functions exists in most all
types of human cancer cells. The ubiquitin-proteasome system
attaches ubiquitin to a target protein, and this process requires
the participation of three core enzymes: ubiquitin activating
enzyme (E1 enzyme), ubiquitin conjugating enzyme (E2 enzyme) and
ubiquitin ligase (E3 ligase) (21).
Generally, ubiquitin tagged proteins are then recognized and
degraded by 26S proteasomes (21).
The ubiquitin-proteasome system controls the turn-over of
regulatory proteins involved in pivotal biological processes
(22,23) and helps modulate the pathological
state of various human diseases (24). Notably, proteasome inhibitors have
shown marked therapeutic benefits in many types of hematological
malignancies (25,26). Therefore, investigation of the
relationship between lncRNAs and ubiquitin-proteasome system in
GCB-DLBCL appears to be valuable. The tumor suppressor gene
p53 has been regarded as ‘the guardian of the genome’, as it
is necessary for the maintenance of genomic stability, and its
inactivation is highly associated with most cancers. However
orchestrating transcription-dependent and -independent cell death
programs, p53 gene plays important roles in cell cycle
progression, as it regulates G1/S, S and G2/M cell cycle
checkpoints (27). The cell cycle
progression is driven by cyclin/cyclin-dependent kinases (CDKs)
including cyclin D/CDK4, cyclin E/CDK2 and cyclin A/CDK2 complexes,
which contribute to the phosphorylation of tumor suppressors of the
RB family, leading to DNA replication (28).
Subsequently, we constructed the lncRNA-mRNA
correlation network that displayed 5 deregulated lncRNAs and their
co-expressed coding genes. In the co-expression network, 3 cell
cycle-related genes including G2 and S phase expressed 1
(GTSE1), cyclin-dependent kinase 4 (CDK4) and
cyclin-dependent kinase 1 (CDK1), which had involvement in
the p53 signaling pathway in the present study, were found to be
negatively correlated with 2 downregulated lncRNAs (ENST00000464929
and ENST00000475089) in our profiles. Furthermore, these 2 lncRNAs
displayed significant negative correlations with 4 pivotal genes in
the ubiquitin-proteasome pathway, including proteasome subunit α7
(PSMA7), proteasome 26S subunit, non-ATPase 14
(PSMD14), ubiquitin conjugating enzyme E2 K (UBE2K)
and ubiquitin conjugating enzyme E2 C (UBE2C). These
findings indicated that ENST00000464929, ENST00000475089 and their
co-expressed coding genes in the ubiquitin-proteasome pathway and
the p53 signaling pathway probably play a significantly collective
role in the pathogenesis of GCB-DLBCL.
Further investigation of the lncRNA-gene network
revealed an association between the levels of ENST00000425358 and
NR_026892 expression and certain carcinogenic or anti-carcinogenic
genes such as the MYB proto-oncogene (MYB), RAS oncogene
family-like 3 (RABL3), and XIAP associated factor 1
(XAF1). These findings highlight the critical roles exerted
by these two upregulated lncRNAs in GCB-DLBCL pathogenesis.
ENST00000425358, also known as HOXA transcript antisense RNA,
myeloid-specific 1 (HOTAIRM1), is a long intergenic non-coding RNA
located at the 3′-end of the HOXA cluster, which is upregulated
during the maturation of myeloid cells (29). HOTAIRM1 is thought to regulate the
expression of numerous genes that determine cell fate. HOTAIRM1 was
previously demonstrated to be specifically expressed in myeloid
lineages of hematopoietic cells (29), but recently HOTAIRM1 was shown to be
overexpressed in cases of basal-like breast cancer (30) and pancreatic ductal adenocarcinoma
(31). Taken together, we inferred
that this long intergenic non-coding RNA may participate in the
occurrence and malignant progression of GCB-DLBCL.
In conclusion, we performed a detailed examination
of lncRNA expression in GCB-DLBCL cells and identified numerous
lncRNAs that displayed aberrant expression in GCB-DLBCL when
compared with normal control. Additionally, we demonstrated that
several differentially expressed lncRNAs were correlated with
multiple Gene Ontology items and pathways involved in
carcinogenesis, suggesting a pivotal role of lncRNAs in the
pathogenesis of GCB-DLBCL. From a clinical point of view, the
potential values of lncRNAs for diagnosis and prognosis prediction
have been well demonstrated (32–34).
In the present study, we presented several candidate lncRNAs which
could serve as biological markers for GCB-DLBCL. However, this
possibility requires more sufficient information concerning
prognosis and cohort study of patients in our subsequent
research.
Glossary
Abbreviations
Abbreviations:
|
lncRNAs
|
long non-coding RNAs
|
|
GCB
|
germinal center B-cell
|
|
DLBCL
|
diffuse large B-cell lymphoma
|
|
qRT-PCR
|
quantitative reverse transcription
polymerase chain reaction
|
|
NHL
|
non-Hodgkin lymphoma
|
|
EZH2
|
enhancer of zeste homolog 2
|
|
ncRNAs
|
non-coding RNAs
|
|
miRs
|
microRNAs
|
|
RLN
|
reactive lymph node
|
|
WHO
|
World Health Organization
|
|
DMEM
|
Dulbecco's modified Eagles medium
|
|
FBS
|
fetal bovine serum
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
DE
|
differentially expressed
|
|
PCC
|
Pearson correlation coefficient
|
|
CDKs
|
cyclin-dependent kinases
|
|
GTSE1
|
G2 and S phase expressed 1
|
|
CDK4
|
cyclin-dependent kinase 4
|
|
CDK1
|
cyclin-dependent kinase 1
|
|
PSMA7
|
proteasome subunit α7
|
|
PSMD14
|
proteasome 26S subunit, non-ATPase
14
|
|
UBE2K
|
ubiquitin-conjugating enzyme E2 K
|
|
UBE2C
|
ubiquitin-conjugating enzyme E2 C
|
|
MYB
|
MYB proto-oncogene
|
|
RABL3
|
RAS oncogene family-like 3
|
|
XAF1
|
XIAP associated factor 1
|
|
HOTAIRM1
|
HOXA transcript antisense RNA,
myeloid-specific 1
|
References
|
1
|
Rodriguez-Abreu D, Bordoni A and Zucca E:
Epidemiology of hematological malignancies. Ann Oncol. 18:(Suppl
1). i3–i8. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alizadeh AA, Eisen MB, Davis RE, Ma C,
Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al:
Distinct types of diffuse large B-cell lymphoma identified by gene
expression profiling. Nature. 403:503–511. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shaffer AL III, Young RM and Staudt LM:
Pathogenesis of human B cell lymphomas. Annu Rev Immunol.
30:565–610. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iqbal J, Sanger WG, Horsman DE, Rosenwald
A, Pickering DL, Dave B, Dave S, Xiao L, Cao K, Zhu Q, et al:
BCL2 translocation defines a unique tumor subset within the
germinal center B-cell-like diffuse large B-cell lymphoma. Am J
Pathol. 165:159–166. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kramer MH, Hermans J, Wijburg E, Philippo
K, Geelen E, van Krieken JH, de Jong D, Maartense E, Schuuring E
and Kluin PM: Clinical relevance of BCL2, BCL6, and MYC
rearrangements in diffuse large B-cell lymphoma. Blood.
92:3152–3162. 1998.PubMed/NCBI
|
|
6
|
Lenz G, Wright GW, Emre NC, Kohlhammer H,
Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al:
Molecular subtypes of diffuse large B-cell lymphoma arise by
distinct genetic pathways. Proc Natl Acad Sci USA. 105:13520–13525.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pasqualucci L, Trifonov V, Fabbri G, Ma J,
Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, et
al: Analysis of the coding genome of diffuse large B-cell lymphoma.
Nat Genet. 43:830–837. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Morin RD, Johnson NA, Severson TM, Mungall
AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et
al: Somatic mutations altering EZH2 (Tyr641) in follicular and
diffuse large B-cell lymphomas of germinal-center origin. Nat
Genet. 42:181–185. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kowalczyk MS, Higgs DR and Gingeras TR:
Molecular biology: RNA discrimination. Nature. 482:310–311. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jardin F and Figeac M: MicroRNAs in
lymphoma, from diagnosis to targeted therapy. Curr Opin Oncol.
25:480–486. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Han L, Zhang K, Shi Z, Zhang J, Zhu J, Zhu
S, Zhang A, Jia Z, Wang G, Yu S, et al: LncRNA profile of
glioblastoma reveals the potential role of lncRNAs in contributing
to glioblastoma pathogenesis. Int J Oncol. 40:2004–2012.
2012.PubMed/NCBI
|
|
12
|
Yang F, Zhang L, Huo XS, Yuan JH, Xu D,
Yuan SX, Zhu N, Zhou WP, Yang GS, Wang YZ, et al: Long non-coding
RNA high expression in hepatocellular carcinoma facilitates tumor
growth through enhancer of zeste homolog 2 in humans. Hepatology.
54:1679–1689. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tahira AC, Kubrusly MS, Faria MF, Dazzani
B, Fonseca RS, Maracaja-Coutinho V, Verjovski-Almeida S, Machado MC
and Reis EM: Long non-coding intronic RNAs are differentially
expressed in primary and metastatic pancreatic cancer. Mol Cancer.
10:1412011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al: Long
non-coding RNA HOTAIR reprograms chromatin state to promote
cancer metastasis. Nature. 464:1071–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McCleland ML, Mesh K, Lorenzana E, Chopra
VS, Segal E, Watanabe C, Haley B, Mayba O, Yaylaoglu M, Gnad F, et
al: CCAT1 is an enhancer-templated RNA that predicts BET
sensitivity in colorectal cancer. J Clin Invest. 126:639–652. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li LJ, Chai Y, Guo XJ, Chu SL and Zhang
LS: The effects of the long non-coding RNA MALAT-1 regulated
autophagy-related signaling pathway on chemotherapy resistance in
diffuse large B-cell lymphoma. Biomed Pharmacother. 89:939–948.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Trimarchi T, Bilal E, Ntziachristos P,
Fabbri G, Dalla-Favera R, Tsirigos A and Aifantis I: Genome-wide
mapping and characterization of Notch-regulated long non-coding
RNAs in acute leukemia. Cell. 158:593–606. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Doose G, Haake A, Bernhart SH, López C,
Duggimpudi S, Wojciech F, Bergmann AK, Borkhardt A, Burkhardt B,
Claviez A, et al: ICGC MMML-Seq Consortium: MINCR is a MYC-induced
lncRNA able to modulate MYC's transcriptional network in Burkitt
lymphoma cells. Proc Natl Acad Sci USA. 112:E5261–E5270. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sehgal L, Mathur R, Braun FK, Wise JF,
Berkova Z, Neelapu S, Kwak LW and Samaniego F: FAS-antisense 1
lncRNA and production of soluble versus membrane Fas in B-cell
lymphoma. Leukemia. 28:2376–2387. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou M, Zhao H, Xu W, Bao S, Cheng L and
Sun J: Discovery and validation of immune-associated long
non-coding RNA biomarkers associated with clinically molecular
subtype and prognosis in diffuse large B cell lymphoma. Mol Cancer.
16:162017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ciechanover A: Proteolysis: From the
lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol.
6:79–87. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hochstrasser M: Ubiquitin, proteasomes,
and the regulation of intracellular protein degradation. Curr Opin
Cell Biol. 7:215–223. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ciechanover A: The ubiquitin-proteasome
proteolytic pathway. Cell. 79:13–21. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ciechanover A: The ubiquitin-proteasome
pathway: On protein death and cell life. EMBO J. 17:7151–7160.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chang JE, Peterson C, Choi S, Eickhoff JC,
Kim K, Yang DT, Gilbert LA, Rogers ES, Werndli JE, Huie MS, et al:
VcR-CVAD induction chemotherapy followed by maintenance rituximab
in mantle cell lymphoma: A Wisconsin Oncology Network study. Br J
Haematol. 155:190–197. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
O'Connor OA: Marked clinical activity of
the proteasome inhibitor bortezomib in patients with follicular and
mantle-cell lymphoma. Clin Lymphoma Myeloma. 6:191–199. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stegh AH: Targeting the p53 signaling
pathway in cancer therapy - the promises, challenges and perils.
Expert Opin Ther Targets. 16:67–83. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sherr CJ: G1 phase progression: Cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang X, Lian Z, Padden C, Gerstein MB,
Rozowsky J, Snyder M, Gingeras TR, Kapranov P, Weissman SM and
Newburger PE: A myelopoiesis-associated regulatory intergenic
non-coding RNA transcript within the human HOXA cluster. Blood.
113:2526–2534. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Su X, Malouf GG, Chen Y, Zhang J, Yao H,
Valero V, Weinstein JN, Spano JP, Meric-Bernstam F, Khayat D, et
al: Comprehensive analysis of long non-coding RNAs in human breast
cancer clinical subtypes. Oncotarget. 5:9864–9876. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou Y, Gong B, Jiang ZL, Zhong S, Liu XC,
Dong K, Wu HS, Yang HJ and Zhu SK: Microarray expression profile
analysis of long non-coding RNAs in pancreatic ductal
adenocarcinoma. Int J Oncol. 48:670–680. 2016.PubMed/NCBI
|
|
32
|
Shao Y, Ye M, Jiang X, Sun W, Ding X, Liu
Z, Ye G, Zhang X, Xiao B and Guo J: Gastric juice long non-coding
RNA used as a tumor marker for screening gastric cancer. Cancer.
120:3320–3328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu M, Xing LQ and Liu YJ: A three-long
non-coding RNA signature as a diagnostic biomarker for
differentiating between triple-negative and non-triple-negative
breast cancers. Medicine. 96:e62222017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma KX, Wang HJ, Li XR, Li T, Su G, Yang P
and Wu JW: Long non-coding RNA MALAT1 associates with the malignant
status and poor prognosis in glioma. Tumour Biol. 36:3355–3359.
2015. View Article : Google Scholar : PubMed/NCBI
|