Introduction
Breast cancer is one of the most common neoplasms
and the leading cause of cancer-related deaths in women worldwide
(1). Unfortunately, almost all
factors known with certainty to cause this disease cannot be easily
prevented, such as genetics and the age at which a woman has a
child. Therefore novel preventive and therapeutic approaches based
on new molecular targets are warranted. Epidemiological studies
indicate that obesity and high-fat diets are considered to be risk
factors and associated with poor prognosis in breast cancer
patients, independently of menopausal status, tumor stage, and
hormonal status (2,3).
Moreover, as is known, cancer cells exhibit
metabolic alterations characterized by increased glycolysis and
lipogenesis, which meet the need of macromolecules and energy to
support rapid growth and proliferation of cancer cells. The most
important and fundamental role of FAs is as building blocks for
newly-synthesized membrane phospholipids. In addition, FAs can also
be used to produce protumorigenic signaling lipids and be used for
mitchondrial β-oxidation to produce ATP (4). Thus, we assumed that blocking the
sources of FA acquisition has the potential to starve cancer cells
and induce apoptosis.
Cancer cells present not only increased de
novo lipid biosynthesis but also modified membrane lipid
composition. Monounsaturated fatty acids (MUFAs) represent
important precursors that form complex lipids including
phospholipids, cholesterol esters, and glycerides, which are the
main component of membranes. Thus, a suitable balance of saturated
fatty acids (SFAs), the end-product of de novo FA synthesis
(5) and MUFAs is critical for
membrane composition affecting membrane fluidity, signal
transduction and gene expression (6). Stearoyl-CoA desaturase 1 (SCD1) is a
critical enzyme which catalyzes the conversion of SFAs into MUFAs.
Recent evidence suggests that the expression of SCD1 is aberrantly
increased in many types of cancer including lung, colon and renal
carcinoma relative to the corresponding normal tissues (6,7), and
SCD1 inhibition has been shown to attenuate cancer cell growth
(8). However, recent studies
revealed that the cytotoxic effects caused by FA synthesis
inhibition can be reversed by exogenous FA supplementation. This
indicates that aside from de novo FA synthesis, FA transport
and uptake are indeed an important and underappreciated aspect of
lipid metabolism in cancer. Furthermore, in the anatomy of the
mammary gland, adipocytes represent one of the most prominent cell
types, thus, cancerous breast glands are embedded in the mammary
fat pad (9). Mammary adipocytes
store and secrete FAs, adipokines, and have the potential to
influence neighboring cells by paracrine and endocrine mechanisms.
Mammary adipocytes appear capable of translocating stored lipids to
breast cancer cells as another key source of FAs (9,10).
Well then, how are FAs transferred from adipocytes to cancer cells?
Evidence shows that FAs especially long-chain fatty acids (LCFAs)
are actively transported across the cell membrane by specialized
proteins instead of passive diffusion (11). The protein-mediated import of LCFAs
is of greatest significance when the metabolic requirements for
LCFAs are high or when the level of FFAs is low (12). Although, several proteins have been
implicated in facilitating FA uptake, CD36 is the best
characterized as an FA translocase (FAT) which enhances LCFA uptake
by overexpression or translocation from intracellular stores to the
plasma membrane (13). Accordingly,
we hypothesized that besides de novo lipogenesis, breast
cancer cells can also uptake exogenous FAs via the transmembrane
channel FAT/CD36, which was found to be overexpressed in the
majority of breast cancer tissues in our study. The therapeutic
efforts aimed to starve cancer cells to death thus suppressing both
FA synthesis and uptake pathways.
In this study, we investigated the role of SCD1 and
CD36 in tumor viability by pharmacologic inhibition or genetic
expression silencing. Our results revealed that breast cancer cells
are highly dependent on the activity of SCD1 in the absence of
exogenous MUFA. Moreover, the data demonstrated that breast cancer
cells can also uptake exogenous MUFA via CD36. Inhibition of both
SCD1 and CD36 resulted in significant antitumor synergy in breast
cancer. Collectively, these results strongly suggest that SCD1 and
CD36 represent viable targets for the development of novel
anticancer agents.
Materials and methods
Materials
MCF-7 human breast cancer cell line was acquired
from the American Type Culture Collection (ATCC). Normal human skin
fibroblasts were obtained from the Laboratory of Clinical Research
Center in Hebei General Hospital. Small molecule SCD1 inhibitor
MF-438 was purchased from Merck Millipore (catalog #569406,
Darmstadt, Germany). Oleic acid and palmitate acid were obtained
from Sigma-Aldrich (catalog #O1383, St. Louis, MO, USA). FA-free
bovine serum albumin (BSA) was from Equitech-Bio (catalog #BAH66,
Kerrville, TX, USA). CellTiter 96 AQueous One Solution cell
proliferation assay was purchased from Promega (MTS; catalog
#G3580, Madison, WI, USA). Hoechst 33342 staining kit was obtained
from Coolaber (catalog #SL7130, Beijing, China).
Cell culture
MCF-7 cells and normal human skin fibroblasts were
cultured in RPMI-1640 medium supplemented with 10% fetal bovine
serum (FBS) (both from Hyclone, Logan, Utah, USA), 100 U/ml
streptomycin, 100 U/ml penicillin at 37°C, 5% CO2, and
100% humidity. For assays, MCF-7 cells were incubated in RPMI-1640
medium with 2% FBS for compound treatment and siRNA treatment.
Small molecule inhibitor and fatty
acid treatment
MF-438 was dissolved in dimethyl sulfoxide (DMSO;
Sigma-Aldrich). Oleic acid and palmitate acid were dissolved in 75%
ethanol to a concentration of 100 mM, then diluted in
phosphate-buffered saline (PBS) containing 10% FA-free BSA
(Equitech-Bio). Both FA-BSA (or BSA) and MF-438 (or DMSO) were
pre-diluted in assay culture medium. For experiments of FA
supplementation, FA bound to BSA (ratio 2:1) was added to the media
at a final concentration of 100 µM.
RNA interference
siRNA duplexes targeting human CD36
(5′-GGAAAGUCACUGCGACAUG-3′) (14)
were synthesized, as well as the negative control (scr)
(5′-UUCUCCGAACGUGUCACGUTT-3′) and were both obtained both from
Sigma-Aldrich. MCF-7 cells were transfected with siRNA at a final
concentration of 100 nM using Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA) according to the manufacturer's protocol. The
transfection efficiency was confirmed by real-time PCR and western
blot analysis.
Cell growth assays
For MF-438 IC50 determinations, MCF-7
cells were plated in 96-well plates at a density of 8,000 or 10,000
cells/well in 10 or 2% FBS, respectively for 24 h. Then cells were
treated with MF-438 or DMSO control only. After 48-h treatment with
the inhibitor, the cell viability was assessed using an MTS assay
according to the manufacturer's protocol. The percentage of
inhibition for wells treated with MF-438 was determined relative to
DMSO alone. FA-BSA was added to the media at 100, 200 or 300 µM,
and the percentage of inhibition with MF-438 was evaluated again.
Then, MCF-7 cells were transfected with CD36-siRNA and the
aforementioned steps were repeated.
Cell apoptosis and death analysis
Apoptosis morphology was observed by fluorescence
microscope (DMI3000B; Leica, Germany) after Hoechst 33342 staining.
Briefly, exponentially growing MCF-7 cells or cells transfected
with CD36-siRNA were seeded into a 24-well plate (1×105
cells/well) for 24 h. Then cells were treated with MF-438 at
approximately the IC50 concentration or DMSO in the
absence or presence of 100 µM of oleic acid for 48 h. The
supernatant was discarded and adhered cells were exposed to Hoechst
33342 at 37°C in dark for 20 min. Finally all specimens were
observed under fluorescence microscope. Apoptosis cells were
identified as cells with condensed and fragmented nuclei. Cell
apoptosis and the death rate were determined by propidium iodide
staining (PI; Sigma-Aldrich) using flow cytometry (FACSCalibur;
Becton Dickinson; BD Biosciences, Franklin Lakes, NJ, USA). MCF-7
cells transfected with CD36-siRNA or not were seeded in a 25-cm
culture flask for 24 h. Then adhered and floating cells were
collected 48 h after MF-438 treatment or DMSO control with or
without 100 µM of oleic acid. PI (1 mg/ml) was added to the cells
for 10 min. Positive PI-stained cells were considered as late
apoptosis and dead cells.
Cell cycle analysis
MCF-7 cells or cells transfected with CD36-siRNA
were treated with MF-438 or DMSO for 48 h in the absence or
presence of 100 µM of oleic acid. At the end of the incubation
period, the cells were trypsinized and fixed with 70% ethanol, then
resuspended in PI solution (50 µg/ml of PI, 100 µg/ml of RNase A
and 0.2% Triton X-100) for 30 min at room temperature. The cell
cycle distribution was determined using a flow cytometer.
Cell scratch test
MCF-7 cells or cells transfected with CD36-siRNA
were plated uniformly in 6-well plates with 80–90% confluence in
culture medium containing 2% FBS. Scratches were made at the bottom
of the plates using the tip of a 200-µl pipette. The following day
then washed with PBS in order to remove the sloughing cells. Then,
the cells were treated with MF-438 or DMSO control in the absence
or presence of 100 µM of oleic acid. Images of the scratches were
acquired using a microscope after 24 and 48 h. The scratch closure
rate was assessed using image processing software (Image J; NIH,
USA). The area between the cells was assessed from 6–8 different
regions on a single scratch.
Oil red O staining
In order to stain intracellular lipid deposits,
MCF-7 cells or cells transfected with CD36-siRNA were cultured on
coverslips in 6-well plates with 100 µM of oleic acid for 48 h.
Then, the cells were fixed with 4% paraformaldehyde and stained
with Oil Red O (Sigma-Aldrich). Images were acquired using a
microscope.
RNA isolation and quantitative
real-time RT-PCR
Total RNA was extracted from cells using standard
TRIzol (Invitrogen) for RNA isolation. The RNA concentration was
determined by assessing the absorbance of a diluted sample at 260
nm using a UV spectrometer (ND 2000; Thermo Fisher Scientific,
Waltham, MA, USA) method. Reverse transcription of RNA was carried
out according to the instructions of the EasyScript First-Strand
cDNA Synthesis SuperMix (Beijing TransGen Biotech Co., Ltd.,
Beijing, China). Quantitative PCR reactions were performed on an
ABI PRISM 7300 instrument (Applied Biosystems Life Technologies,
Foster City, CA, USA) using SYBR-Green I GoTaq® qPCR
Master Mix (Promega). PCRs were carried out in a total of 20 µl as
follows: one cycle at 95°C for 5 min, followed by 40 cycles of 95°C
for 30 sec, 58°C for 30 sec and 72°C for 30 sec. Then the PCR
products were analyzed by melting curve to confirm the specificity
of amplification. The gene expression from each sample was analyzed
in triplicate and GAPDH was used as an internal control. The
results are expressed as the relative gene expression using the
ΔΔCt method. Specific primers designed for amplification of CD36
(forward, 5′-CGGAACTGTGGGCTCAT-3′ and reverse,
5′-GGTCTCCAACTGGCATTAGAA-3′) and GAPDH (forward,
5′-GGATGATGTTCTGGAGAGCC-3′ and reverse, 5′-CATCACCATCTTCCAGGAGC-3′)
were verified by NCBI BLAST database.
Western blot analysis
Cells were washed twice with ice-cold PBS and
scraped into 1 ml of RIPA lysis buffer (Sangon Biotech Co., Ltd.,
Shanghai, China) with protease inhibitors of PMSF. Lysates were
clarified by centrifugation (12,000 rpm for 10 min) and the
supernatants were collected. Equal amounts of protein (50 µg)
underwent SDS-PAGE and then were electrotransferred to PVDF
membranes (Millipore Corp., Billerica, MA, USA), which were then
sealed at room temperature for 2 h. The membranes were then
incubated overnight at 4°C with the following primary antibodies
diluted in blocking buffer: SCD1 (catalog sc-14715), and CD36
(catalog sc-7309) (both from Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), and the internal control β-actin (Cell Signaling
Technology, Inc. Danvers, MA, USA). Then, the membranes were washed
and incubated with the appropriate HRP-conjugated secondary
antibody (Beijing CoWin Bioscience Co., Ltd., Beijing, China) in
PBST for 2 h at room temperature. The membranes were then washed
three times and reacted with chemiluminescent agent for 5 min. Then
they were ECL labeled, exposed, and displayed. Quantification of
the resulting images was performed by densitometry with Gel-Pro
Analyzer 4.0 software (Media Cybernetics, Inc., Bethesda, MD, USA)
and the final readings were normalized against β-actin.
Construction of tissue microarray and
immunohistochemistry
The clinical study was approved by the Ethics
Committee of Hebei General Hospital. Sixty-five breast cancer
specimens from resected breast tissue were obtained at the Hebei
General Hospital between 2010–2013 following patient informed
consent. A tissue microarray (TMA) of breast cancer (65 cases) and
adjacent normal breast tissues (37 cases) was prepared by our team
manually. A representative area was selected from an H&E
section and the corresponding area was marked on the surface of the
paraffin-embedded breast cancer tissue block. Then, a paraffin
tissue punch was used to extract a 1.8-mm core sample from the
selected area, which was placed into a recipient block and linked
to a database that contained clinicopathological data. TMA sections
(4 µm) were stained using standard immunohistochemistry (IHC)
techniques for expression and localization of SCD1 and CD36
according to the supplier's protocol. ImmPRESS anti-goat or
anti-mouse peroxidase polymer detection systems along with a
NovaRED kit as a substrate were used for the peroxidase-mediated
reaction.
Statistical analysis
All experiments were repeated at least three times.
Values are represented as the means ± SD. Statistical analyses were
performed with the SPSS statistical package (SPSS 16.0 software).
Statistical significance was assessed by ANOVA (post-hoc used the
Student-Newman-Keuls method) and unpaired Student's t-tests.
P<0.05 was considered to indicate a statistically significant
difference.
Results
SCD1 inhibition results in a serum
dependent decrease of cell proliferation in MCF-7 cells
MF-438 is a small molecule that specifically
inhibits SCD1 enzymatic activity. To determine the effects of
MF-438 on proliferation, MCF-7 cells were treated with MF-438 from
100 nmol/l to 100 µmol/l and the cell viability was assessed after
48 h. MF-438 revealed a significant dose-dependent proliferation
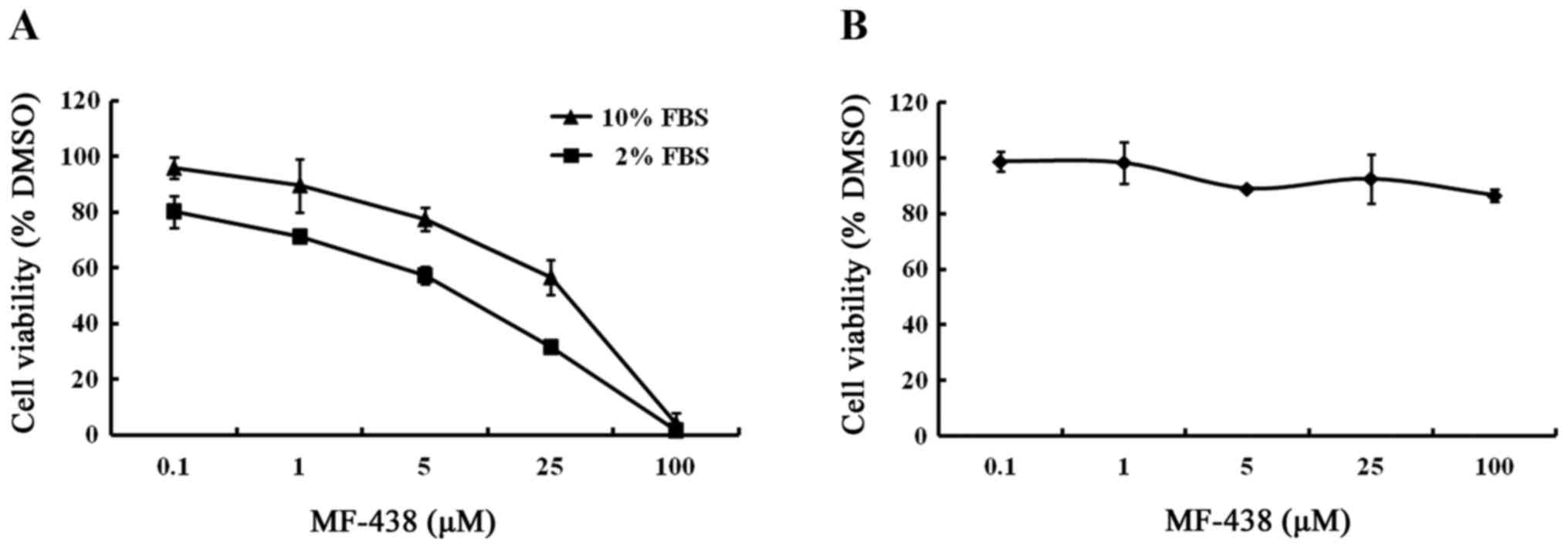
inhibition in MCF-7 cells. Fig. 1A
shows concentration response curves of MCF-7 cells treated in 10 or
2% FBS conditions. Under the 10% FBS condition, the IC50
value was determined to be 16.7 µmol/l, whereas the IC50
in 2% FBS was 3.7 µmol/l. The growth viability of MCF-7 cells was
only slightly slowed when cultured in 2% FBS without compound
treatment relative to the 10% serum concentration. These results
revealed that cells grown in decreased FBS were more sensitive to
growth inhibition of the SCD1 inhibitor.
SCD1 inhibition does not impair the
proliferation of normal human fibroblasts
To investigate the influence of the SCD1 inhibitor
on normal cells, human fibroblasts were incubated for 48 h, enough
time to ensure at least one population doubling, with MF-438 at
concentrations ranging from 100 nmol/l to 100 µmol/l in medium
containing 10% FBS. As displayed in Fig. 1B, the SCD inhibitor did not impair
the growth of normal human fibroblasts. This demonstrates that
normal cells have much less requirement for endogenously produced
MUFA than cancer cells.
MUFA rescue cell proliferation
impaired by SCD1 inhibition in MCF-7 cells
To confirm that MCF-7 cells sensitive to SCD1
inhibition in decreased serum conditions are related to limiting
availability of MUFAs, we investigated whether the anti-growth
effects of the SCD1 inhibitor could be rescued by the addition of
exogenous FA. MCF-7 cells were incubated with 5 µM of MF-438 for 48
h in the presence of either MUFA or SFA. Addition of BSA alone did
not alter the dose response of cells to MF-438. However,
supplementation with oleic acid-BSA revealed a dose-dependent
rescue of cell viability to MF-438 treatment. To extend this
result, we also assessed the effects of supplementing media with
SFA palmitic acid, the substrate of SCD1. The results revealed that
far from protecting cells from growth inhibition by the SCD1
inhibitor, addition of palmitic acid produced a modest viability
decrease at the concentrations used (Fig. 2A). The cellular IC50
values for growth inhibition by MF-438 were determined in different
conditions and are shown in Table
I. These results indicate that the major viability impact
observed in SCD1 inhibition is due to depletion of MUFAs, the
downstream products of SCD1, while on the other hand, part of this
effect may be induced by the resulting accumulation of SFAs, the
upstream substrates of SCD1. SCD1 activity or MUFAs protect cells
against cytotoxicity of SFAs.
 | Table I.IC50 values for the
inhibition of the cell viability by MF-438 in MCF-7 cells cultured
in media with 10 or 2% FBS, and various concentrations of oleic
acid or palmitic acid added to 2% FBS. |
Table I.
IC50 values for the
inhibition of the cell viability by MF-438 in MCF-7 cells cultured
in media with 10 or 2% FBS, and various concentrations of oleic
acid or palmitic acid added to 2% FBS.
| Culture media | IC50
(µM) of MF-438 |
|---|
| 2% FBS | 3.7 |
| 10% FBS | 16.7 |
| 100 µM oleic
acid | 20.0 |
| 200 µM oleic
acid | 32.2 |
| 300 µM oleic
acid | 38.5 |
| 100 µM palmitate
acid | 0.3 |
CD36 depletion results in a
non-MUFA-dependent decrease in breast cancer cell
proliferation
Both the expression of CD36 mRNA (24 h after
transfection) and protein (48 h after transfection) were markedly
suppressed in CD36-depleted MCF-7 cells (Fig. 3). Then, these cells were incubated
with 5 µM of MF-438 or DMSO vehicle for 48 h in the absence or
presence of 100 µM of oleic acid. As shown in Fig. 2B, MF-438 treatment also exhibited a
dose-dependent inhibition of cell proliferation. CD36-depletion
treatment slightly decreased cell proliferation compared with
normal MCF-7 cells, in other words, CD36 depletion increased the
cell sensitivity to MF-438. However, supplementation with oleic
acid did not exhibit an obvious rescue effect of cell viability to
MF-438 in CD36-depleted MCF-7 cells. This revealed that the
anti-proliferative effects of MF-438 could not be reversed by
exogenous oleic acid when the CD36 gene was depleted.
Oleic acid rescues the cell apoptosis
induced by SCD1 inhibition
To further examine the anti-proliferation effects of
MF-438 on apoptosis, MCF-7 cells were treated with 5 µM of MF-438
for 48 h. Hoechst staining revealed that the apoptotic MCF-7 cells
were stained bright blue fluorescence while the normal cells were
stained slightly blue (Fig. 4A).
The typical apoptotic morphology was observed in most MCF-7 cells
treated with MF-438 including cell shrinkage, chromatin
condensation, marginalization and fragmentation as well as the
apoptotic bodies. Furthermore, the result of PI staining indicated
that the rate of late apoptotic and dead cells was significantly
higher in the MF-438 treated group (4.78±0.22%) than the DMSO
control (0.54±0.12%, p=0.000) (Fig.
4B).
We then supplied SCD1-inhibited MCF-7 cells with 100
µM of oleic acid. It was observed that the apoptotic cells were
significantly decreased after oleic acid addition. The result of PI
staining revealed that exogenous oleic acid alleviated cytotoxicity
and decreased the rate of cell death caused by SCD1 inhibition
(1.06±0.35% for oleic acid combined with MF-438 vs. 4.78±0.22% for
MF-438 alone, p=0.000) (Fig. 4B).
Thus, we demonstrated that the pharmacological SCD1
inhibitor-induced apoptotic cell death can be rescued by exogenous
oleic acid.
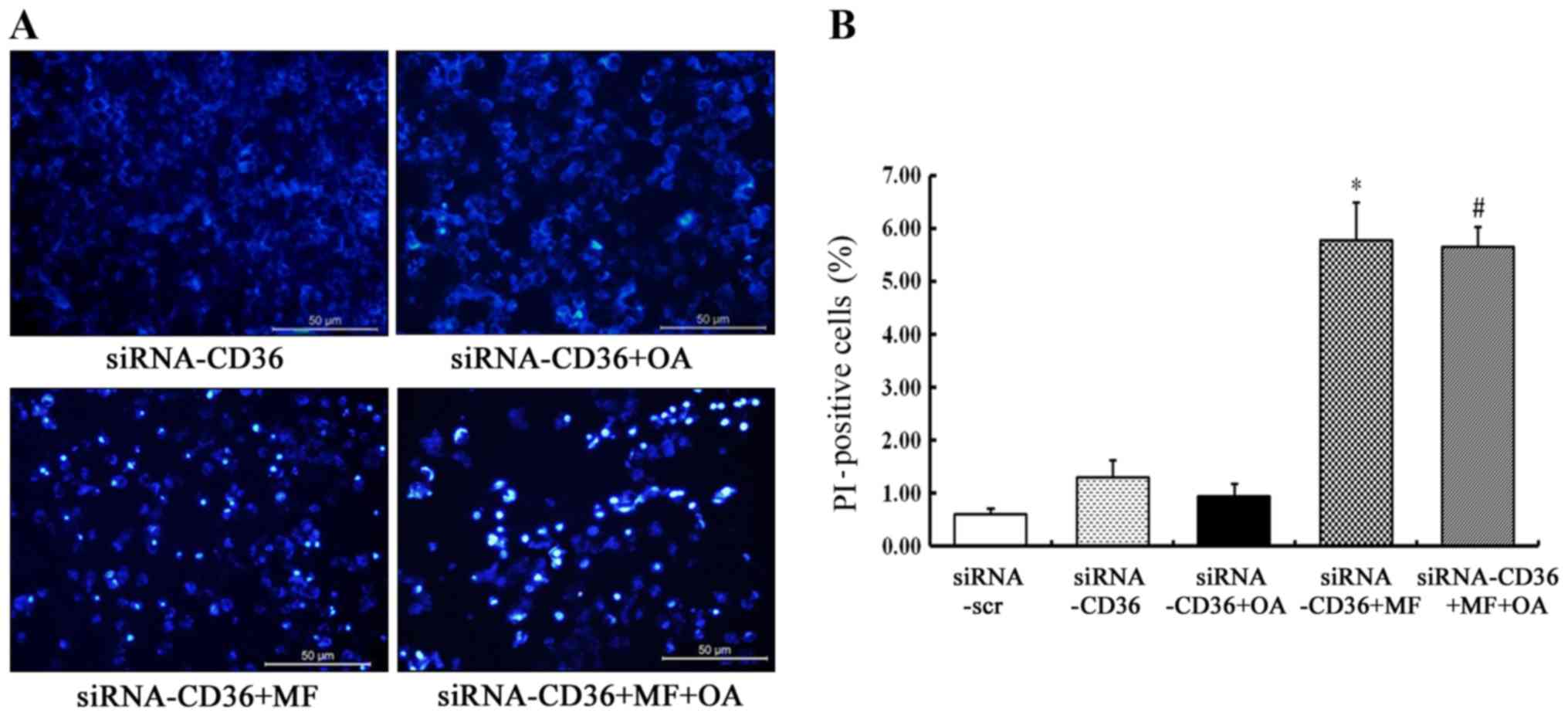
CD36 depletion prevents the rescue
effect of oleic acid on SCD1 inhibition-induced cell apoptosis
CD36-silenced MCF-7 cells were incubated with 5 µM
of MF-438 or DMSO for 48 h in the absence or presence of 100 µM of
oleic acid. The typical apoptotic morphology was found in most
CD36-depleted MCF-7 cells treated with MF-438 (Fig. 5A). PI staining revealed that the
rate of late apoptotic and dead cells was significantly higher when
CD36-silenced MCF-7 cells were treated with MF-438 (5.77±0.72% for
MF-438 vs. 1.30±0.33% for siRNA-CD36 control, p=0.000) whether
oleic acid was added or not (5.65±0.38% for oleic acid combined
with MF-438 vs. 5.77±0.72% for MF-438 alone, p=0.998) (Fig. 5B). The result demonstrated that the
rescue effect of oleic acid on the SCD1 inhibitor-induced cell
apoptosis was exerted only if CD36 was present.
Oleic acid reverses the cell cycle
blocked by SCD1 inhibition
To better understand the mechanism involved in
growth inhibition by the SCD1 inhibitor, MCF-7 cells were incubated
with 5 µM of MF-438 in serum-decreased media for 48 h and the cell
cycle distribution was analyzed by flow cytometry. The results
revealed a significant decrease in the population of cells in the S
phase (35.3 vs. 44.8%, respectively, p=0.001) and the G2/M phase
(2.6 vs. 4.9%, respectively, p=0.003) with MF-438 treatment
compared to the DMSO control (Fig.
6A). A concomitant increase in the percentage of cells in the
G0/G1 phase was also observed (62.1 vs. 50.4%, respectively,
p=0.001). This indicated that SCD1 inhibition specifically targeted
the progression of the cell cycle at the level of the synthetic
phase.
However, when 100 µM of oleic acid-BSA was
supplemented in the aforementioned cells, the population of cells
in the S phase (42.1 vs. 44.8%, respectively, p=0.315), G2/M phase
(4.8 vs. 4.9%, p=1.0) and the G0/G1 phase (53.1 vs. 50.4%, p=0.466)
were all reversed in the MF-438-treated group compared to the DMSO
control (Fig. 6A). Oleic acid
reversed the changes of the cell cycle caused by the SCD1
inhibitor, suggesting that the lipid components of serum, possibly
MUFA, were able to ensure the SCD1 inhibition of cells through
mitosis.
CD36 depletion prevents the rescue
effect of oleic acid on the restoration of the cell cycle arrest of
SCD1 inhibited cells
CD36-depleted MCF-7 cells were also incubated with
MF-438 for 48 h in the absence or presence of oleic acid. Fig. 6B also revealed a significant
decrease in the population of CD36-depleted cells in the S phase
(24.0 vs. 40.5%, respectively, p=0.000) and the G2/M phase (1.7 vs.
6.4%, respectively, p=0.001) with MF-438 treatment compared to the
siRNA-CD36 control, and a concomitant increase in the percentage of
cells in the G0/G1 phase (74.4 vs. 53.2%, respectively, p=0.000).
Oleic acid supplementation did not alter the cell cycle profile,
with the percentage of S phase 25.3vs. 24.0% (p=0.370), G2/M phase
2.0 vs. 1.7% (p=0.986), and G0/G1 phase 72.7 vs. 74.4% (p=0.357),
respectively in οleic acid combined with MF-438 treatment compared
to MF-438 treatment alone (Fig.
6B). The results revealed that the cell cycle arrest caused by
the SCD1 inhibitor cannot be reversed by exogenous oleic acid when
the CD36 gene was silenced.
Oleic acid restores the migration
ability decreased by SCD1 inhibition
The results (Fig.
7A) revealed that the scratches of the cells treated with
MF-438 were wider than the DMSO control at the same time-point
(30.52±0.73 vs. 18.47±0.90 µm at 24 h P=0.000; 22.80±1.10 vs.
12.70±1.04 µm at 48 h P=0.000). However, when oleic acid was added
to the cells treated with MF-438, the scratches became
significantly narrower compared to MF-438 group (27.81±0.81 vs.
30.52±0.73 µm at 24 h P=0.000; 17.18±1.25 vs. 22.80±1.10 µm at 48 h
P=0.000) (Fig. 7B). This indicated
that the SCD1 inhibitor was able to suppress migration in MCF-7
cell line, however the addition of oleic acid restored the
migration ability.
CD36 depletion prevents the rescue
effect of oleic acid on the restoration of the migratory ability of
SCD1 inhibited cells
The scratches of the CD36-silenced MCF-7 cells
treated with MF-438 were wider than the DMSO control (38.30±0.77
vs. 35.20±0.91 µm at 24 h P=0.002; 38.14±0.66 vs. 25.47±1.27 µm at
48 h P=0.000). When oleic acid was supplemented, there were no
obvious changes in the width of the scratches compared to
MF-438-treated group (37.02±1.17 vs. 38.30±0.77 µm at 24 h P=0.685;
36.67±0.33 vs. 38.14±0.66 µm at 48 h P=0.545) (Fig. 8). Exogenous oleic acid did not
reverse the migration ability in CD36-depleted MCF-7 cells.
Overall, these results indicated that blockade in
SCD1 activity could not enable MCF-7 cells to get through the early
stages of the cell cycle, leading to cell apoptosis and making
cells migrate more slowly. Exogenous oleic acid markedly reversed
the effect caused by SCD1 inhibition but only if CD36 was
present.
Breast cancer cells can use exogenous
FAs for tumor growth
To further determine whether breast cancer cells
also uptake exogenous FAs via CD36 as an alternative pathway to
obtain lipids, we added oleic acid in the media of MCF-7 cells and
CD36-silenced MCF-7 cells. It was observed that more cytoplasmic
lipid droplets accumulated in MCF-7 cells but not in CD36-silenced
MCF-7 cells (Fig. 9) after oleic
acid supplementation.
SCD1 and the CD36 protein are highly
expressed in human breast cancer
IHC was performed to detect SCD1 and the protein
level of CD36 in human breast cancer specimens and adjacent normal
breast tissues. SCD1 exhibited diffuse cytoplasmic staining and was
highly expressed in nearly all breast cancer samples, whereas it
was low or not expressed in adjacent normal breast tissues
(Fig. 10). Similarly, the majority
of breast cancer samples were also stained for CD36 mainly at the
plasma membrane (Fig. 10).
Discussion
Cancer cells are distinct from normal cells based
partly on their unique metabolic status, one aspect of which is an
unusual requirement for FA metabolism to sustain cell division and
proliferation, fulfill energy requirements and provide metabolites
for anabolic processes. Numerous studies have documented that LCFAs
influence the proliferation of cancer cells both in vivo and
in vitro (8). The critical
role of lipids in cancer cell proliferation has led to a number of
proposed strategies for treating cancer through inhibition of lipid
availability.
Thus the FA synthesis pathway has been an attractive
cancer target for quite some time, and attention is primarily
focused on FA synthase with the production of long-chain SFAs
(15). However, MUFAs play a more
important role in dynamic or rapidly dividing cancer cells and are
a major constituent of biological structures such as membranes, and
can also function as or modify signaling molecules. SCD1 is the
critical enzyme in the synthesis of MUFAs, oleic and palmitoleic
acid, from SFA, stearic and palmitic acid. SCD1 may represent a
final rate-limiting point in the FA synthesis pathway. We revealed
in the present study that loss of SCD1 activity yielded pronounced
growth inhibition in breast cancer MCF-7 cells while producing no
notable effects in normal cells. This was also observed in colon
and lung cancer cells in vitro (16,17).
In the present study, we provided new evidence that SCD1 controls
breast cancer cell proliferation through induction of apoptosis,
cell cycle arrest and migration prevention.
Moreover, our data revealed that the
anti-proliferative effect of the SCD1 inhibitor was more sensitive
under low serum conditions. Furthermore, this growth inhibition
could be reversed by oleic acid in all aspects aforementioned but
not palmitic acid which even produced the opposite effect. These
results imply that the major viability impact is attributable to an
SCD1-inhibition-mediated oleic acid deficiency, while on the other
hand, partly due to the buildup of intracellular palmitic acid.
Although it appears that SFA enhanced the anti-proliferative effect
of SCD1 inhibition in MCF-7 cells, human normal cells are more
sensitive to the toxicity of excess SFA than cancer cells which may
cause cardiovascular diseases and fatty liver called lipotoxicity.
SCD1, overexpressed in cancer cells but not normal cells, appears
to be part of a safeguard mechanism for cancer cells to confront
the SFA-mediated toxicity.
While cancer cells are characterized by persistent
cell division, the number of proliferating cells is also affected
by the rate of cell death. The present results indicate that SCD1
is a critical factor for breast cancer cells to promote cell
survival and prevent programmed cell death through its production
of MUFA. Furthermore, our observation that breast cancer cells
treated with the SCD1 inhibitor are arrested in the G1 phase and
that this effect is rescued by oleic acid suggests that MUFA
synthesis is required in the early phases of the cell cycle. In
fact, we have known for years that a high rate of membrane
phospholipid synthesis and turnover occurs during the G1 and early
S phases for cell division (18).
Our results demonstrated that constant activation of SFA synthesis
coordinated with subsequent conversion of SFA into MUFA is required
to provide the phospholipid biosynthesis with MUFA substrates for
new membrane synthesis before or during the synthetic G1/S phase of
the cell cycle. Moreover, the presence of abundant unsaturated
lipids in cancer cells may have critical implications for their
biological phenotype. Highly expressed SCD1 in cancer cells
enriches the membrane phospholipids with MUFA thereby producing a
more fluid lipid membrane, which is thought to induce growth
factor-activated proliferation, migration and invasion (19,20).
Provided that these observations may be extended to
the in vivo situation, human breast cancer proliferation
involves a complex interaction between genes, hormones, calorigenic
nutrients and the microenvironment. Obesity which is manifested as
increased circulating FFAs (e.g., oleic acid), and a high fat diet
especially a diet rich in oleate may decrease the antitumor effect
of SCD1 inhibition and favor mammary tumor progression. Conversely,
this indicates that the antitumor effect of the SCD1 inhibitor is
steadily exerted possibly only in low fat diet conditions or non
obese patients. Thus, this may require patients to eat a low fat
diet and control their body weight or body fat.
However, many tumors associated with obesity reside
near anatomic adipose tissue depots, including renal, pancreatic,
colon and breast. For instance, adipocytes are a major component of
the stromal microenvironment of mammary ducts and glands where
breast cancer arises. Emerging data has revealed that adipocytes
participate in the crosstalk with neoplastic cells and promote
cancer initiation and progression, however the mechanisms involving
the paracrine effects of adipokines and adipocyte products
transferred to cancer cells have not been fully clarified yet
(21,22). Some studies have shown that
adipocytes promote tumorigenesis through providing free FAs to
ovarian cancer cells and glutamine to leukemia cells (23,24).
From our aforementioned observations, we realize that lipid
transport and uptake maybe also be an important and
underappreciated aspect of lipid metabolism in breast cancer
especially within an adipocyte-rich environment (23). Thus, the fact that SCD1 inhibition
fails to have constant effects on tumor growth is not surprising
given the ability of cancer cells to uptake unsaturated lipid from
their microenvironment.
Unlike glucose, which is water soluble, exogenous
FAs are either circulating FA-albumin complexes or LPL-mediated
hydrolysis of plasma lipoproteins. CD36, as a transmembrane
protein, binds and concentrates exogenous LCFAs at the membrane,
facilitating their translocation across the plasma membrane.
Previous studies revealed that CD36-deficient mice display changes
in LCFA utilization and metabolism consistent with an important
role for the molecule in LCFA transport (25–27).
The channel protein CD36 mediates the uptake of FAs in some normal
tissues with FA metabolism, including adipose tissue and muscle,
but is expressed at low levels (28). However, our study revealed that CD36
expression is increased in breast cancer samples, and that
cytoplasmic lipid droplets accumulate only in cells in which CD36
exists after exogenous oleic acid addition. This indicates that
breast cancer cells may obtain exogenous FAs via CD36 from the
microenvironment (e.g., adipocytes) or circulating particles (e.g.,
diet-derived lipoprotein particles). Thus, alterations in CD36
expression could influence the uptake of exogenous FAs. Our results
also confirm that the antitumor effect caused by SCD1 inhibitor can
not be reversed by exogenous oleic acid when CD36 gene is depleted.
This study provides the first evidence of the effect of CD36 in
exogenous FA uptake by breast cancer cells, making CD36 an ideal
candidate for therapeutic intervention in patients presenting with
breast cancer.
In summary, SCD1 may control the overall rate of
lipid synthesis and then affect cell proliferation in breast cancer
cells. SCD1 may be a promising molecular target for cancer therapy.
Consistent with FA synthesis, FA uptake and transport may be
another important target pathway for anticancer therapy. Our study
suggests that there was a synergistic cytostatic effect when both
pathways were blocked simultaneously. Selective inhibition of key
enzymes in endogenous lipogenesis, such as SCD1, combined with an
exogenous FA uptake channel protein, such as CD36, may be a
potential chemotherapeutic strategy for breast cancer. Although,
our investigations reveal a synergistic antitumor response in
breast cancer cells to SCD1 inhibition paired with CD36 depletion,
the exact mechanism of this synergistic effect is not well defined,
and further studies must be conducted in some other cell lines.
Acknowledgements
This study was supported by the Natural Science
Foundation of Hebei Province, China (grant no. H2016307004).
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Majed B, Moreau T, Senouci K, Salmon RJ,
Fourquet A and Asselain B: Is obesity an independent prognosis
factor in woman breast cancer? Breast Cancer Res Treat.
111:329–342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lorincz AM and Sukumar S: Molecular links
between obesity and breast cancer. Endocr Relat Cancer. 13:279–292.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rysman E, Brusselmans K, Scheys K,
Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D,
Daniëls VW, Machiels J, et al: De novo lipogenesis protects cancer
cells from free radicals and chemotherapeutics by promoting
membrane lipid saturation. Cancer Res. 70:8117–8126. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Igal RA: Stearoyl-CoA desaturase-1: A
novel key player in the mechanisms of cell proliferation,
programmed cell death and transformation to cancer. Carcinogenesis.
31:1509–1515. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
von Roemeling CA, Marlow LA, Wei JJ,
Cooper SJ, Caulfield TR, Wu K, Tan WW, Tun HW and Copland JA:
Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target
for clear cell renal cell carcinoma. Clin Cancer Res. 19:2368–2380.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hess D, Chisholm JW and Igal RA:
Inhibition of stearoylCoA desaturase activity blocks cell cycle
progression and induces programmed cell death in lung cancer cells.
PLoS One. 5:e113942010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hovey RC and Aimo L: Diverse and active
roles for adipocytes during mammary gland growth and function. J
Mammary Gland Biol Neoplasia. 15:279–290. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kuemmerle NB, Rysman E, Lombardo PS,
Flanagan AJ, Lipe BC, Wells WA, Pettus JR, Froehlich HM, Memoli VA,
Morganelli PM, et al: Lipoprotein lipase links dietary fat to solid
tumor cell proliferation. Mol Cancer Ther. 10:427–436. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stremmel W, Pohl L, Ring A and Herrmann T:
A new concept of cellular uptake and intracellular trafficking of
long-chain fatty acids. Lipids. 36:981–989. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schaffer JE: Fatty acid transport: The
roads taken. Am J Physiol Endocrinol Metab. 282:E239–E246. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Krammer J, Digel M, Ehehalt F, Stremmel W,
Füllekrug J and Ehehalt R: Overexpression of CD36 and acyl-CoA
synthetases FATP2, FATP4 and ACSL1 increases fatty acid uptake in
human hepatoma cells. Int J Med Sci. 8:599–614. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ozdener MH, Subramaniam S, Sundaresan S,
Sery O, Hashimoto T, Asakawa Y, Besnard P, Abumrad NA and Khan NA:
CD36- and GPR120-mediated Ca2+ signaling in human taste
bud cells mediates differential responses to fatty acids and is
altered in obese mice. Gastroenterology. 146:995–1005. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kuhajda FP: Fatty acid synthase and
cancer: New application of an old pathway. Cancer Res.
66:5977–5980. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mason P, Liang B, Li L, Fremgen T, Murphy
E, Quinn A, Madden SL, Biemann HP, Wang B, Cohen A, et al: SCD1
inhibition causes cancer cell death by depleting mono-unsaturated
fatty acids. PLoS One. 7:e338232012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Noto A, Raffa S, De Vitis C, Roscilli G,
Malpicci D, Coluccia P, Di Napoli A, Ricci A, Giovagnoli MR,
Aurisicchio L, et al: Stearoyl-CoA desaturase-1 is a key factor for
lung cancer-initiating cells. Cell Death Dis. 4:e9472013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jackowski S: Cell cycle regulation of
membrane phospholipid metabolism. J Biol Chem. 271:20219–20222.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stambolic V and Woodgett JR: Functional
distinctions of protein kinase B/Akt isoforms defined by their
influence on cell migration. Trends Cell Biol. 16:461–466. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ge G, Wu J and Lin Q: Effect of membrane
fluidity on tyrosine kinase activity of reconstituted epidermal
growth factor receptor. Biochem Biophys Res Commun. 282:511–514.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mathur A, Hernandez J, Shaheen F, Shroff
M, Dahal S, Morton C, Farrior T, Kedar R and Rosemurgy A:
Preoperative computed tomography measurements of pancreatic
steatosis and visceral fat: Prognostic markers for dissemination
and lethality of pancreatic adenocarcinoma. HPB Oxf. 13:404–410.
2011. View Article : Google Scholar
|
|
22
|
Muller C: Tumour-surrounding adipocytes
are active players in breast cancer progression. Ann Endocrinol
(Paris). 74:108–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nieman KM, Kenny HA, Penicka CV, Ladanyi
A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB,
Hotamisligil GS, et al: Adipocytes promote ovarian cancer
metastasis and provide energy for rapid tumor growth. Nat Med.
17:1498–1503. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ehsanipour EA, Sheng X, Behan JW, Wang X,
Butturini A, Avramis VI and Mittelman SD: Adipocytes cause leukemia
cell resistance to L-asparaginase via release of glutamine. Cancer
Res. 73:2998–3006. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Febbraio M, Podrez EA, Smith JD, Hajjar
DP, Hazen SL, Hoff HF, Sharma K and Silverstein RL: Targeted
disruption of the class B scavenger receptor CD36 protects against
atherosclerotic lesion development in mice. J Clin Invest.
105:1049–1056. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Coburn CT, Knapp FF Jr, Febbraio M, Beets
AL, Silverstein RL and Abumrad NA: Defective uptake and utilization
of long chain fatty acids in muscle and adipose tissues of CD36
knockout mice. J Biol Chem. 275:32523–32529. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Febbraio M, Guy E, Coburn C, Knapp FF Jr,
Beets AL, Abumrad NA and Silverstein RL: The impact of
overexpression and deficiency of fatty acid translocase (FAT)/CD36.
Mol Cell Biochem. 239:193–197. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Doege H and Stahl A: Protein-mediated
fatty acid uptake: Novel insights from in vivo models. Physiology
(Bethesda). 21:259–268. 2006. View Article : Google Scholar : PubMed/NCBI
|