Introduction
Cell contact inhibition is a critical property of
normal cells to stop cell proliferation upon reaching confluence
(1). In vitro,
non-transformed cells are arrested in G0/G1 phase at high density
to form a confluent monolayer. In vivo, contact inhibition
keeps homeostasis of cell number and thus maintains normal tissue
architecture and the organ size (2). In contrast to normal cells, cancer
cells usually exhibit a loss of growth control and escape contact
inhibition, enabling them to replicate limitlessly (3,4). There
are numbers of oncogenes and tumor suppressor genes reported to
regulate contact inhibition, but the precise mechanisms behind the
loss of cell contact inhibition of cancer cells remain largely
unknown.
The c-myc proto-oncogene encodes a ubiquitously
expressed transcription factor which plays a central role in cell
growth and proliferation regulating (5). It has been reported that c-Myc is
deregulated in nearly half of human tumors, such as hepatocellular
carcinoma (HCC) and cholangiocarcinoma (CCA) (6–8). c-Myc
contains a basic region and a helix-loop-helix/leucine zipper
(HLH/LZ) domain. Through the HLH/LZ domain, c-Myc heterodimerizes
with another transcription factor Max (9,10). The
c-Myc/Max complex activates the expression of target genes through
binding enhancer box sequences (E-boxes) and recruiting histone
acetyltransferases (HATs) (11).
c-Myc is induced rapidly when cells transit from quiescent to
proliferative state, and the induction of c-Myc is required for
cell growth and proliferation (12,13).
It has been reported that forced overexpression of c-Myc can induce
cell cycle re-entry in quiescent cells, and the inhibition of c-Myc
can lead to the suppression of cell cycle (14,15).
CCA is a highly malignant tumor originating from
biliary epithelial cells (16). As
usually diagnosed at advanced stage, the prognosis of CCA is very
poor. CCA is a heterogeneous tumor and the molecular mechanisms
underlying the development of CCA are largely unknown. Since c-Myc
is induced during cholestatic liver injury and CCA progression, and
c-Myc knockdown inhibits the progression of CCA (17,18),
it is important to uncover the detailed mechanism of c-Myc in
CCA.
Owing to the critical role of c-Myc in cell
proliferation regulation, we wonder whether c-Myc participates in
the loss of contact inhibition of CCA cells. Here, we find that
c-Myc sustains high protein levels in confluent human CCA cells,
but not in human normal biliary epithelial cells. The inhibition of
c-Myc restores contact inhibition of human CCA cells. Furthermore,
we show that c-Myc promotes the loss of contact inhibition in human
CCA cells through the mTOR pathway, and the deregulation of c-Myc
is controlled by Yes-associated protein (YAP). Our findings
indicate that c-Myc is an important promoter mediating the loss of
contact inhibition in human CCA cells.
Materials and methods
Chemicals and antibodies
c-Myc inhibitor 10058-F4 (F4) and YAP inhibitor
verteporfin (VP) were purchased from Selleck Chemicals (Houston,
TX, USA). mTOR inhibitor rapamycin was purchased from Tocris
Bioscience (Bristol, UK). Antibodies against cyclin D1, p27, c-Myc,
p-p70S6K, p70S6K, p-S6, p-YAP (ser127), YAP, p-Merlin and Merlin
were purchased from Cell Signaling Technology (Danvers, MA, USA).
Antibody against GAPDH, human c-Myc siRNA and control siRNA were
purchased from Santa Cruz Biotechnology (Heidelberg, Germany).
Cell culture
Human normal biliary epithelial cell line HIBEC and
CCA cell lines QBC939 and RBE were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum and 1%
penicillin/streptomycin in a humidified incubator containing 5%
CO2 and 95% ambient air at 37°C. Cells were plated at
different densities to achieve low (−50% confluent cells) and high
(confluent cells) densities.
Western blot analysis
Cells were lysed in Triton lysis buffer (20 mM Tris,
pH 7.4, 137 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, 1 mM
PMSF, 10 mM NaF, 5 mg/ml aprotinin, 20 mM leupeptin, and 1 mM
sodium orthovanadate) and centrifuged at 12,000 g for 15 min.
Protein concentrations were measured using the BCA assay. Protein
samples were denatured with 4X SDS-loading buffer (200 mM Tris, pH
6.8, 8% SDS, 400 mM DTT, 0.4% bromphenol blue, 40% glycerol) at
100°C for 5 min and subjected to standard SDS-PAGE and western blot
analysis as previously described (19). The proteins were electrolytically
transferred to an NC membrane and blocked in TBST containing 5%
non-fat milk at room temperature for 1 h. The membrane was
incubated at 4°C overnight with primary antibodies against cyclin
D1 (1:1,000), p27 (1:1,000), c-Myc (1:1,000), p-p70S6K (1:1,000),
p70S6K (1:1,000), p-S6 (1:1,000), p-YAP (ser127) (1:1,000), YAP
(1:1,000), p-Merlin (1:1,000), Merlin (1:1,000) and GAPDH (1:200).
Then the membrane was incubated with secondary antibodies labeled
with IRDye 700 (Rockland Immunochemicals, Gilbertsville, PA, USA).
Protein levels were detected by the Odyssey system (LiCor, Lincoln,
NE, USA).
Cell cycle analysis by flow
cytometry
Cells were fixed overnight, resuspended in
phosphate-buffered saline (PBS), and stained with propidium iodide
in the dark for 30 min. The DNA content was measured by
fluorescence-activated cell sorting (FACS) on a Becton-Dickinson
FACScan flow cytometry system.
RNA interference
Transient transfections were performed with
Lipofectamine 2000 (Invitrogen) according to the manufacturer's
instructions. Briefly, for each 35-mm dish, 10 µl of 20 µM stock of
duplex was mixed with 100 µl of DMEM while in a separate tube 8 µl
of Lipofectamine 2000 was mixed with 100 µl of DMEM. The two
solutions were then gently combined and added to prewashed cells,
in normal medium including serum. The cultures were then left for 6
h before washing and refeeding.
Statistical analysis
Results were expressed as the mean ± SD. Statistical
analysis was performed using Student's t-test. P<0.05 was
considered statistically significant.
Results
Human CCA cells are resistant to
contact inhibition
To determine the roles of cell density in the
proliferation of normal biliary epithelial cells and CCA cells, we
plated normal biliary epithelial cells HIBEC and CCA cells QBC939
and RBE at low density and high density. The flow cytometry data
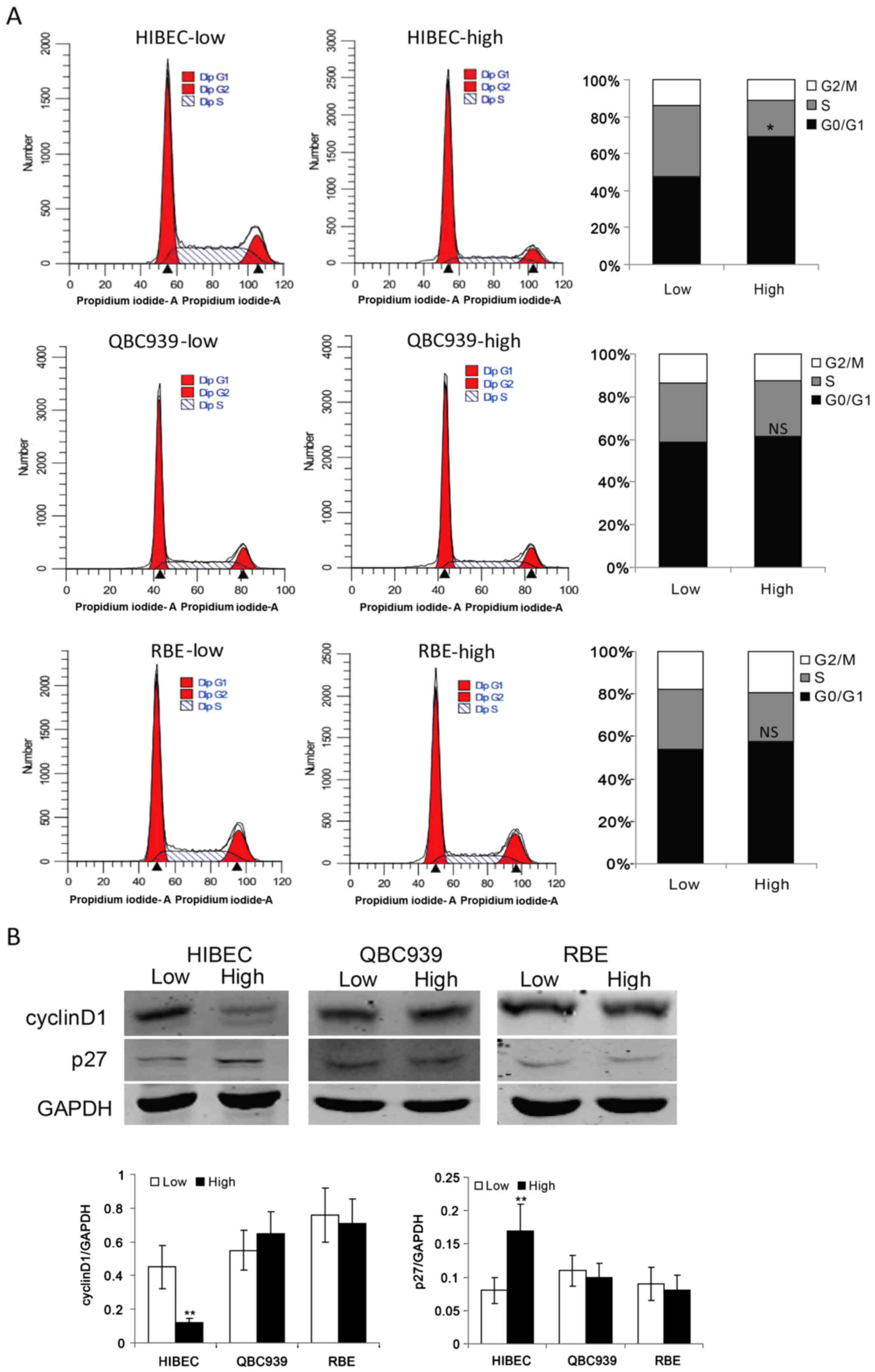
showed that contact-inhibited normal biliary epithelial cells HIBEC
were arrested in G0/G1 phase (Fig.
1A). It is notable that CCA cells QBC939 and RBE keep strong
proliferation ability at high density (Fig. 1A). These data indicated that CCA
cells are resistant to contact inhibition. As cyclin D1, which is
required for G1→S phase transition, is a key regulator of cell
cycle progression (20), we checked
the protein levels of cyclin D1 in HIBEC, QBC939 and RBE cells at
low and high densities, respectively. Compared with low density,
high density obviously decreased the protein level of cyclin D1 in
HIBEC cells (Fig. 1B). While, high
density had no apparent effect on the protein levels of cyclin D1
in QBC939 and RBE cells (Fig. 1B).
p27, a marker of G0/G1 phase arrest, has been shown to play an
important role in contact inhibition (21,22).
Then, we assessed the protein levels of p27 in HIBEC, QBC939 and
RBE cells at low and high densities. As shown in Fig. 1B, contact inhibition induced the
expression of p27 in HIBEC cells. However, in QBC939 and RBE cells,
there was no apparent difference of p27 protein levels between low
and high densities. These data indicate that CCA cells are
resistant to contact inhibition.
c-Myc contributes to contact
inhibition loss in human CCA cells
Since c-Myc is a crucial regulator for cell cycle,
and its deregulation is implicated in CCA (23), we set out to investigate whether
c-Myc is involved in contact inhibition resistance in CCA cells. We
evaluated the protein levels of c-Myc in HIBEC, QBC939 and RBE
cells at low and high densities. We found that c-Myc protein levels
are clearly lower in confluent HIBEC cells as compared with low
density HIBEC cells (Fig. 2A).
However, there was no apparent difference in the protein levels of
c-Myc between low and high densities in QBC939 and RBE cells
(Fig. 2A). c-Myc protein maintained
high levels in confluent QBC939 and RBE cells (Fig. 2A). These data indicate that c-Myc
may be an important promoter for CCA cells to escape contact
inhibition. To determine whether c-Myc leads to contact inhibition
resistance in CCA cells, confluent QBC939 and RBE cells were
treated with c-Myc specific inhibitor 10058-F4. Our data are
consistent with other studies that 10058-F4 effectively suppress
the expression of c-Myc (Fig. 2B).
Furthermore, the data demonstrated that cyclin D1 clearly decreased
upon 10058-F4 treatment in confluent QBC939 and RBE cells (Fig. 2B).
We detected the cell cycle in
confluent CCA cells after 10058-F4 treatment
The results showed that 10058-F4 treatment induced
G0/G1 phase cell cycle arrest in confluent QBC939 and RBE cells
(Fig. 3A). To confirm the role of
c-Myc in inhibiting contact inhibition, we used siRNA to knock-down
the expression of c-Myc. As shown in Fig. 3B, c-Myc knockdown induced G0/G1
phase cell cycle arrest in confluent QBC939 and RBE cells. These
data suggest that c-Myc is required for CCA cells to override
contact inhibition.
The mTOR pathway is implicated in
c-Myc-mediated contact inhibition loss in human CCA cells
How c-Myc induces contact inhibition loss in CCA
cells was next investigated. It has been reported that the
inactivation of mammalian target of rapamycin (mTOR) is implicated
in cell contact inhibition (24).
We have previously reported that the mTOR pathway plays an
important role in CCA cell growth and survival (25). Here we wonder whether there is a
link between c-Myc and mTOR in contact inhibition regulation in CCA
cells. We investigated the activity of mTOR in HIBEC and CCA cells
at different cell densities. The results showed the levels of
phosphorated p70S6K and S6 ribosomal protein (S6), both are markers
of the activity of mTOR, decreased obviously in confluent HIBEC
cells. In contrast, there is no apparent decrease of
phosphor-p70S6K and phosphor-S6 in confluent QBC939 and RBE cells
(Fig. 4A). In order to determine
whether there is a link between c-Myc and mTOR in confluent CCA
cells, we investigated the activity of mTOR in confluent CCA cells
upon c-Myc inhibition. The results showed that inhibition of c-Myc
by 10058-F4 significantly suppressed the phosphorylation of p70S6K
and S6 in confluent QBC939 and RBE cells (Fig. 4B). To further confirm the results,
we used siRNA to knock down c-Myc expression. As shown in Fig. 4C, c-Myc knockdown suppressed the
phosphorylation of p70S6K and S6 as well as the protein levels of
cyclin D1 in confluent QBC939 and RBE cells. These data suggest
that the mTOR pathway is involved in c-Myc-induced contact
inhibition loss in CCA cells.
To confirm whether mTOR is required for the loss of
contact inhibition in CCA cells, we detected cell cycle status in
confluent CCA cells after mTOR inhibition. As shown in Fig. 5, mTOR inhibitor rapamycin treatment
induced G0/G1 arrest in confluent QBC939 and RBE cells. Together,
these data indicate that c-Myc/mTOR deregulation plays a pivotal
role in contact inhibition loss in CCA cells.
Deregulated c-Myc in confluent human
CCA cells is regulated by YAP signaling
Next, we investigated the underlying mechanism of
c-Myc deregulation in confluent CCA cells. As Yes-associated
protein (YAP) plays an important role in regulating cell contact
inhibition and in cancer development (26,27).
We tested whether YAP is involved in c-Myc-mediated contact
inhibition loss in CCA cells. The levels of phospho-YAP and total
YAP proteins at low and high densities in HIBEC, QBC939 and RBE
cells were examined. The data showed that phospho-YAP (ser127), an
inactive form of YAP, markedly increased in HIBEC cells at high
density (Fig. 6A), implying
increased YAP protein was sequestrated in cytoplasm and was
inactivated. In QBC939 and RBE cells, there was no apparent
increase of the levels of phospho-YAP (ser127) at high density
(Fig. 6A), indicating YAP sustains
high levels of activity in confluent CCA cells. To explore whether
c-Myc is regulated by YAP, we detected c-Myc protein levels after
YAP inhibition by a specific inhibitor verteporfin in CCA cells. As
shown in Fig. 6B, the protein
levels of c-Myc significantly decreased in confluent CCA cells upon
verteporfin treatment. YAP inhibition decreased cyclin D1 in
confluent CCA cells. These data demonstrate that YAP is involved in
c-Myc-mediated contact inhibition loss in CCA cells.
As Merlin (also named neurofibromatosis type 2 gene,
NF2) is a pivotal inhibitor of YAP (26,28),
we investigated the activities of Merlin in HIBEC, QBC939 and RBE
cells at low and high densities. The results showed that high
density induced decrease of phosphor-Merlin in HIBEC cells
(Fig. 6C), indicating that the
upregulation of Merlin activity is required for contact inhibition
in normal biliary epithelial cells. It is notable that the levels
of phosphor-Merlin increased in confluent QBC939 and RBE cells
(Fig. 6C), indicating that the
activity of Merlin is downregulated in confluent CCA cells.
Together, these data imply a potential role of Merlin/ YAP/c-Myc
signaling axis for CCA cells to override contact inhibition.
Discussion
Strict control of normal tissue size is achieved by
regulating cell numbers. In normal tissues, cell number is
controlled by the mechanism known as cell contact inhibition.
Contact inhibition is a crucial mechanism to regulate cell
proliferation in vivo and in vitro. Loss of contact
inhibition is a hallmark of most human cancer cells (4). As a result, cancer cells can maintain
the ability of proliferation upon reaching confluence. This
criterion is also commonly used for cellular transformation in
vitro (3). Consistently, we
found normal biliary epithelial cells that showed apparent growth
suppression at high density, while the proliferation ability of CCA
cells were not apparently affected by high density. Although some
important players, such as the Hippo pathway, which mediate cell
cycle arrest in contact inhibition have been identified (27), the detailed molecular mechanisms
mediating contact inhibition loss in cancer cells remain largely
unknown.
Although the deregulation of c-Myc plays an
important role in the pathogenesis of CCA (29), detailed mechanisms of how c-Myc
promotes CCA remain largely unknown. Both normal biliary epithelial
cells and human CCA cells showed strong expression of c-Myc at low
density. Importantly, c-Myc clearly decreased in confluent normal
biliary epithelial cells, but not in confluent CCA cells. As high
density induced growth arrest of normal biliary epithelial cells,
and CCA cells still sustained high ability of proliferation at high
density, it is reasonable to suggest that c-Myc might be involved
in contact inhibition loss in CCA cells. This speculation is
supported by our data which demonstrated that the blockade of c-Myc
by inhibitor 10058-F4 effectively restores contact inhibition in
confluent CCA cells. Thus, c-Myc can help CCA cells to override
cell-cell contact inhibition and promote the progress of CCA.
The mTOR pathway is a central pathway that regulates
cell growth, survival and metabolism (30). The mTOR pathway deregulation
contributes to the development and progression of most human
cancers (31). Importantly, it has
been reported that the deactivation of mTOR signaling is an
important event for cell contact inhibition (24). Consistently, our data show that the
activity of mTOR decreased obviously in contact-inhibited normal
biliary epithelial cells. We and others have previously found that
the mTOR pathway is implicated in the carcinogenesis and
progression of CCA and mTOR inhibition suppresses the growth of
human CCA cells (25,32). Whether the implication of mTOR in
CCA is associated with contact inhibition regulation has not been
investigated. Here, our data show that CCA cells at low and high
densities both display strong activity of mTOR. Based on the data
that blocking mTOR by rapamycin initiated G0/G1 cell cycle arrest
in confluent CCA cells, we suggest that mTOR promotes CCA cells to
override contact inhibition. Importantly, we find that c-Myc
suppression obviously decreased the activity of mTOR in confluent
CCA cells. Thus, c-Myc promotes the loss of contact inhibition
through the mTOR pathway in CCA cells. Further studies are needed
to investigate the association between c-Myc and mTOR in CCA.
As the c-Myc protein decreased at confluence status
in normal biliary epithelial cells but not in CCA cells, a major
issue now is how c-Myc sustains high protein levels in confluent
CCA cells. It has been widely reported that the oncoprotein YAP
plays an essential role in cell contact inhibition and normal
tissue growth control (26).
Accordantly, our data show that the activity of YAP is suppressed
upon high density in normal biliary epithelial cells but not in CCA
cells. To figure out whether the deregulated expression of c-Myc is
associated with YAP in confluent CCA cells, YAP inhibitor
verteporfin, a small molecule that inhibits TEAD-YAP association,
was used in our studies. The results show that verteporfin
effectively decreased the protein levels of c-Myc and cyclin D1 in
confluent CCA cells. Our results are consistent with the findings
by Turato et al which demonstrated that knockdown YAP by
siRNA reduced c-Myc proteins (33).
These data demonstrated that the deregulated c-Myc in confluent CCA
cells is regulated by YAP signaling. YAP is a central effector in
the Hippo pathway, controlling cell-cell contact inhibition
(27). It is well known that Merlin
is an upstream regulator of the Hippo pathway (27,28).
Our data show that Merlin is involved in the deregulation of YAP in
confluent CCA cells, and the deactivation of Merlin in confluent
CCA cells plays an important role in YAP/c-Myc/mTOR signaling
axis-mediated contact inhibition loss. It has been reported that
Merlin-deficient nervous system tumors show loss of contact
inhibition and activation of Wnt/β-catenin and its downstream c-Myc
(34). Further studies are needed
to clarify whether Wnt signaling is implicated in the
Merlin-YAP-c-Myc in our model, and to uncover the detailed
mechanisms of Merlin deregulation in confluent CCA cells.
In conclusion, this study presents a new mechanism
in which Merlin/YAP/c-Myc/mTOR signaling axis promotes human CCA
cell proliferation by overriding contact inhibition. More detailed
studies on the mechanism of Merlin aberrant inactivation and the
link between c-Myc and mTOR in CCA cells will contribute to the
understanding of molecular mechanism of cholangiocarcinogenesis and
the development of new therapeutic strategies against CCA.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (81472312), Innovation Team of
Education Department of Sichuan Province (16TD0021), Science and
Technology Department of Sichuan Province Foundation (2017JY0134),
Luzhou City-Southwest Medical University Foundation
(2016LZXNYD-T02, 2013LZLY-J06, 2015LZCYD-S01-14/15 and
2015LZCYD-S01-8/15), Sichuan Province-Luzhou City-Southwest Medical
University Foundation (14JC0082, 14JC0038 and 14ZC0070) and
Education Department of Sichuan Province Foundation (15ZA0160 and
17ZA0437).
References
|
1
|
Eagle H and Levine EM: Growth regulatory
effects of cellular interaction. Nature. 213:1102–1106. 1967.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carter SB: Tissue homeostasis and the
biological basis of cancer. Nature. 220:970–974. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abercrombie M: Contact inhibition and
malignancy. Nature. 281:259–262. 1979. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brodeur GM, Seeger RC, Schwab M, Varmus HE
and Bishop JM: Amplification of N-myc in untreated human
neuroblastomas correlates with advanced disease stage. Science.
224:1121–1124. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kaposi-Novak P, Libbrecht L, Woo HG, Lee
YH, Sears NC, Coulouarn C, Conner EA, Factor VM, Roskams T and
Thorgeirsson SS: Central role of c-Myc during malignant conversion
in human hepatocarcinogenesis. Cancer Res. 69:2775–2782. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang H, Liu T, Wang J, Li TW, Fan W, Peng
H, Krishnan A, Gores GJ, Mato JM and Lu SC: Deregulated methionine
adenosyltransferase α1, c-Myc, and Maf proteins together promote
cholangiocarcinoma growth in mice and humans. Hepatology.
64:439–455. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Follis AV, Hammoudeh DI, Daab AT and
Metallo SJ: Small-molecule perturbation of competing interactions
between c-Myc and Max. Bioorg Med Chem Lett. 19:807–810. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu J, Banerjee A and Goss DJ: Assembly of
b/HLH/z proteins c-Myc, Max, and Mad1 with cognate DNA: Importance
of protein-protein and protein-DNA interactions. Biochemistry.
44:11855–11863. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park S, Chung S, Kim KM, Jung KC, Park C,
Hahm ER and Yang CH: Determination of binding constant of
transcription factor myc-max/max-max and E-box DNA: The effect of
inhibitors on the binding. Biochim Biophys Acta. 1670:217–228.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dean M, Levine RA, Ran W, Kindy MS,
Sonenshein GE and Campisi J: Regulation of c-myc transcription and
mRNA abundance by serum growth factors and cell contact. J Biol
Chem. 261:9161–9166. 1986.PubMed/NCBI
|
|
13
|
Reed JC, Alpers JD, Nowell PC and Hoover
RG: Sequential expression of protooncogenes during
lectin-stimulated mitogenesis of normal human lymphocytes. Proc
Natl Acad Sci USA. 83:3982–3986. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schuhmacher M and Eick D: Dose-dependent
regulation of target gene expression and cell proliferation by
c-Myc levels. Transcription. 4:192–197. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Mannava S, Grachtchouk V, Zhuang
D, Soengas MS, Gudkov AV, Prochownik EV and Nikiforov MA: c-Myc
depletion inhibits proliferation of human tumor cells at various
stages of the cell cycle. Oncogene. 27:1905–1915. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Blechacz BR and Gores GJ:
Cholangiocarcinoma. Clin Liver Dis. 12:131–150, ix. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang H, Li TW, Ko KS, Xia M and Lu SC:
Switch from Mnt-Max to Myc-Max induces p53 and cyclin D1 expression
and apoptosis during cholestasis in mouse and human hepatocytes.
Hepatology. 49:860–870. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang H, Li TW, Peng J, Tang X, Ko KS, Xia
M and Aller MA: A mouse model of cholestasis-associated
cholangiocarcinoma and transcription factors involved in
progression. Gastroenterology. 141(378–388): 388e371-374. 2011.
|
|
19
|
Zhao X, Fu J, Xu A, Yu L, Zhu J, Dai R, Su
B, Luo T, Li N, Qin W, et al: Gankyrin drives malignant
transformation of chronic liver damage-mediated fibrosis via the
Rac1/JNK pathway. Cell Death Dis. 6:e17512015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Quelle DE, Ashmun RA, Shurtleff SA, Kato
JY, Bar-Sagi D, Roussel MF and Sherr CJ: Overexpression of mouse
D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes
Dev. 7:1559–1571. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Motti ML, Califano D, Baldassarre G,
Celetti A, Merolla F, Forzati F, Napolitano M, Tavernise B, Fusco A
and Viglietto G: Reduced E-cadherin expression contributes to the
loss of p27kip1-mediated mechanism of contact inhibition in thyroid
anaplastic carcinomas. Carcinogenesis. 26:1021–1034. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bretones G, Delgado MD and León J: Myc and
cell cycle control. Biochim Biophys Acta. 1849:506–516. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leontieva OV, Demidenko ZN and
Blagosklonny MV: Contact inhibition and high cell density
deactivate the mammalian target of rapamycin pathway, thus
suppressing the senescence program. Proc Natl Acad Sci USA.
111:8832–8837. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao X, Zhang C, Zhou H, Xiao B, Cheng Y,
Wang J, Yao F, Duan C, Chen R, Liu Y, et al: Synergistic antitumor
activity of the combination of salubrinal and rapamycin against
human cholangiocarcinoma cells. Oncotarget. 7:85492–85501.
2016.PubMed/NCBI
|
|
26
|
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim
J, Xie J, Ikenoue T, Yu J, Li L, et al: Inactivation of YAP
oncoprotein by the Hippo pathway is involved in cell contact
inhibition and tissue growth control. Genes Dev. 21:2747–2761.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zeng Q and Hong W: The emerging role of
the hippo pathway in cell contact inhibition, organ size control,
and cancer development in mammals. Cancer Cell. 13:188–192. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang N, Bai H, David KK, Dong J, Zheng Y,
Cai J, Giovannini M, Liu P, Anders RA and Pan D: The Merlin/NF2
tumor suppressor functions through the YAP oncoprotein to regulate
tissue homeostasis in mammals. Dev Cell. 19:27–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Berasain C, Fernández-Barrena MG and Avila
MA: New molecular interactions of c-Myc in cholangiocarcinoma may
open new therapeutic opportunities. Hepatology. 64:336–339. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li X, Wu C, Chen N, Gu H, Yen A, Cao L,
Wang E and Wang L: PI3K/Akt/mTOR signaling pathway and targeted
therapy for glioblastoma. Oncotarget. 7:33440–33450. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim LC, Cook RS and Chen J: mTORC1 and
mTORC2 in cancer and the tumor microenvironment. Oncogene.
36:2191–2201. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yothaisong S, Dokduang H, Techasen A,
Namwat N, Yongvanit P, Bhudhisawasdi V, Puapairoj A, Riggins GJ and
Loilome W: Increased activation of PI3K/AKT signaling pathway is
associated with cholangiocarcinoma metastasis and PI3K/mTOR
inhibition presents a possible therapeutic strategy. Tumour Biol.
34:3637–3648. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Turato C, Cannito S, Simonato D, Villano
G, Morello E, Terrin L, Quarta S, Biasiolo A, Ruvoletto M, Martini
A, et al: SerpinB3 and Yap interplay increases Myc oncogenic
activity. Sci Rep. 5:177012015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou L, Ercolano E, Ammoun S, Schmid MC,
Barczyk MA and Hanemann CO: Merlin-deficient human tumors show loss
of contact inhibition and activation of Wnt/β-catenin signaling
linked to the PDGFR/Src and Rac/PAK pathways. Neoplasia.
13:1101–1112. 2011. View Article : Google Scholar : PubMed/NCBI
|