Introduction
In addition to its anti-inflammatory and analgesic
actions, the antitumor function of parthenolide (PT), a
sesquiterpene lactone extracted from the plant feverfew, has been
widely studied in recent years. For example, Sun et al
demonstrated that PT inhibited proliferation and induced autophagy
in HepG2 cells by suppressing the expression of Ki67, a
proliferation-related gene. Moreover, PT has been demonstrated to
promote apoptosis in hepatoma carcinoma cells by downregulating
Bcl-2 and upregulating p53 and Bax (1). PT also decreased the viability of
MDA-MB-231 breast cancer cells. Inhibition of MDA-MB-231 cells by
PT may be associated with superoxide anion and highly reactive
oxygen species (ROS) (2). Studies
have also revealed that PT inhibited SiHa and MCF-7 breast cancer
cells in a concentration-dependent manner by upregulating p53, Bax,
caspase-3 and −6 and inhibiting Bcl-2 (3). PT increased ROS, and then induced cell
death, AMPK activation and the cell cycle arrest in breast cancer
cells (4). In addition, Yu et
al reported that in oral cancer cells, PT-induced apoptosis was
impaired by siRNA targeting Bcl-2-interacting mediator of cell
death (BIM). PT also increased the expression of death receptor 5
(DR5), which is closely related to caspase-8-mediated apoptosis.
Thus, PT promoted BIM and DR5 expression and subsequently induced
apoptosis in oral cancer cells to suppress cancer (5). Collectively, these studies revealed
that PT inhibits various tumor cells and solid tumors.
We revealed that PT markedly inhibited the
proliferation and migration of several colorectal cancer cell
lines. However, the half maximal inhibitory concentration
(IC50) of PT in colorectal cancer cells increased during
long-term administration, indicating the development of PT
resistance. As few studies have examined the mechanism underlying
PT resistance, we focused on this resistance in the present study.
To this end, we selected 3 colorectal cancer cell lines, HCT-116,
HT-29 and Caco-2, to establish PT-resistant cell lines using the
concentration gradient-increased induction method and screened for
differentially expressed genes in the resistant cells and their
parental cells using gene arrays. Bioinformatic analysis and data
summary significantly revealed lower expression of Smad4 in the
resistant cells than in the parental cells, which was confirmed by
subsequent evaluation of Smad4 mRNA and protein levels in the
resistant cells and parental cells (data not shown). The literature
search revealed that Smad4 has a direct effect on the
phosphorylation, or the nuclear transport of Smad2 and Smad3, thus,
the change in the expression of Smad4 may affect downstream
proteins in the TGF-β signaling pathway. For these reasons, we
decided to select Smad4 as the target for the present study.
Smad4 is closely associated with cancer: it
primarily acts as a tumor suppressor and is often inactivated in
tumors. Specifically, inactivation or mutation of Smad4 has been
identified during the progression and metastasis of many types of
cancer, including prostate (6),
ovarian (7), colorectal (8) and thyroid (9) cancers. Studies of aberrant Smad4
expression and tumor drug resistance have demonstrated that Smad4
expression affects the chemosensitivity of some tumors to certain
drugs. For example, Boulay et al reported that patients with
colorectal cancer and normal Smad4 diploidy derived a 3-fold higher
benefit from 5-fluorouracil (5-FU)-based adjuvant chemotherapy
(overall survival, 3.23, P=0.056; disease-free survival, 2.89,
P=0.045) than patients with a Smad4 deletion (10). Furthermore, Smad4 deletion or
inactivation promoted colorectal cancer cell proliferation,
migration, invasion and resistance to 5-FU, whereas LY294002, an
inhibitor of the PI3K/AKT pathway, restored sensitivity to 5-FU in
Smad4-deficient cells. Smad4 inactivation induced resistance to
5-FU in HCT-116 Smad4−/− cells (11). These studies all demonstrated the
impact of Smad4 on the sensitivity of colorectal cancer cells to
5-FU. In the present study, we explored the influence of the Smad4
gene on the sensitivity to PT and the possible involvement of Smad4
target proteins in drug resistance. Moreover, we investigated the
efficacy of combined Smad4 gene intervention and PT in the
inhibition of the development of PT-resistant human colorectal
xenografted tumors.
Materials and methods
Cell culture
Three colorectal cancer cell lines, HCT-116, HT-29
and Caco-2, were purchased from the Cell Bank of the Chinese
Academy of Sciences (CCAS; Shanghai, China) and maintained in
RPMI-1640 medium containing 10% fetal bovine serum (FBS) (both from
Invitrogen, Shanghai, China). PT (10 µM) was added to the complete
medium used in the culture of HCT-116/PT, HT-29/PT and Caco-2/PT.
The viral packaging cell line 293TN was purchased from the American
Type Culture Collection (ATCC; Rockville, MD, USA) and maintained
in Dulbeccos minimum essential medium, (DMEM; Invitrogen)
supplemented with 10% FBS. All cells were incubated at 37°C in a
humidified 95% air and 5% CO2 incubator. All adherent
cells were passaged using 0.25% trypsin (Invitrogen) digestion.
Establishment of resistant colorectal
cancer cell lines by induction
HCT-116, HT-29 and Caco-2 cells were maintained
according to routine protocols, except for the addition of PT
(starting at 0.5 µM) (Sigma-Aldrich, Carlsbad, CA, USA) to the
medium; the concentration of PT was doubled every 3 passages, i.e.,
1, 2, 4, 8, 16 and 32 µM. After being cultured in medium containing
32 µM PT for 3 passages, resistant cells were named as HCT-116/PT,
HT-29/PT and Caco-2/PT. We then examined the IC50 values
for PT in the resistant cell lines and their parental cells. The
resistant cells as well as their parental cell lines were seeded
into 96-well plates (Corning, Corning, NY, USA) at a concentration
of 1×105 cells/well and cultured for 48 h in medium
containing PT at final concentrations of 1, 2, 4, 8, 16 or 32 µM.
The Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) assay was
employed to assess the sensitivity of the cells to PT, and the
IC50 values were calculated. Moreover, PT-resistant cell
lines and their parental cell lines (1×106 cells) were
harvested for total RNA and protein extraction, followed by
real-time PCR and western blotting for Smad4 assessment.
Construction of a pcDNA-Smad4
plasmid
The coding sequence (CDS) of human Smad4
(NM_005359.5) was amplified using the primers
5′-GGAATTCGCCACCATGGACAATGTGTCTATTACG-3′ and
5′-CGGGATCCTCAGTCTAAAGGTTGTGGGTCTG-3′, which contained an
EcoRI site, a Kozak sequence and a BamHI site. cDNA
was prepared by reverse transcription using RNA isolated from 293TN
cells. The PCR product was digested and cloned into a pcDH1-CMV
lentiviral expression vector (System Biosciences, Mountain View,
CA, USA); the recombinant vector was named pcDH1-Smad4. The
construct integrity was confirmed by DNA sequencing, and
endotoxin-free DNA was prepared in all cases. The products of the
vectors were confirmed by DNA sequencing, and endotoxin-free DNA
was prepared.
Lentivirus packaging
One day prior to transfection, 293TN cells were
seeded into 10-cm dishes and co-transfected with 2 µg of the
pcDH1-Smad4 vector and 10 µg of pPACK Packaging Plasmid mix (System
Biosciences) using Lipofectamine 2000 (Invitrogen) in accordance
with the manufacturer's protocol. The medium was then replaced with
DMEM containing 1% FBS. After 48 h, the supernatant was harvested
and then cleared by centrifugation for 5 min at 5,000 × g and 4°C
before being passed through a 0.45-µm polyvinylidene fluoride
(PVDF) membrane (Millipore, Billerica, MA, USA). The viral titer
was determined by gradient dilution, and the packaged lentivirus
was named Lv-Smad4. A control virus carrying no insert gene,
Lv-control, was constructed as well.
Overexpression of Smad4 in
PT-resistant colorectal cancer cell lines using a lentiviral
approach
HCT-116/PT, HT-29/PT and Caco-2/PT cells in
logarithmic growth phase were seeded into 6-well plates at
5×105 cells/well, and cultured for 24 h in complete
medium at 37°C and 5%CO2. One day later, a viral
solution (Lv-control or Lv-Smad4) was added at a multiplicity of
infection (MOI) of 10. The infection efficiency was observed using
a fluorescent marker at 72 h after infection. Total RNA and protein
were isolated from the cells and subjected to real-time PCR and
western blotting for evaluation of Smad4 mRNA and protein levels,
respectively.
Assessment of cell viability and
IC50 values
The 3 cell lines and their derivatives were seeded
into 96-well plates at 5×104 cells/well and cultured in
medium containing PT at final concentrations of 1, 2, 4, 8, 16 or
32 µM for 48 h, followed by a CCK-8 assay to assess cell viability.
Briefly, 10 µl of CCK-8 solution was added, and the cells were then
cultured under normal conditions for an additional 4 h before
measuring the absorbance at 450 nm. The inhibition ratio was
calculated, and the fitted equation between the inhibition ratio
and PT concentration was obtained for computation of the
IC50 values for PT at 48 h.
Detection of apoptosis
The resistant cells were seeded into 6-well plates
at 1×105 cells/well in medium containing 10 µM PT and
cultured for 48 h. The cells were collected, and apoptosis was
assessed using flow cytometry (FACSCalibur) after treatment with
Annexin V/FITC apoptosis detection kit II (cat. 556570) (both from
BD Biosciences, San Jose, CA, USA). Each cell line was grouped as
control cells, cells infected with Lv-control and cells infected
with Lv-Smad4, and then cultured in the presence or absence of PT.
For lentiviral infection, cells were employed in the experiment 72
h after infection. The experiments were carried out in 3 resistant
cell lines.
Assessment of cell migration
Cell migration was assessed using a QCM™ 24-well
Fluorimetric Cell Migration Assay kit (Chemicon, Temecula, CA, USA)
in accordance with the manufacturer's instructions. Cells were
seeded into the upper chamber at 1×106 cells/well, and
500 µl of complete medium containing 10% FBS was added to each well
in the lower chamber. Cells that had migrated to the underside of
the membrane were fixed in 4% paraformaldehyde after 48 h. The
invading cells were stained with DAPI, and fluorescence was
quantified. Data are reported as relative fluorescence units
(RFUs). The grouping was the same as that in the apoptosis
assay.
Effects of overexpression of Smad4 on
relevant proteins in PT-resistant colorectal cancer cells
HCT-116/PT, HT-29/PT and Caco-2/PT and those
infected with Lv-Smad4 for 72 h were seeded into 6-well plates at
1×105 cells/well and treated with PT (10 µM) or without
PT. After being cultured at 37°C under 5% CO2 for 48 h,
the cells were harvested, and total protein was extracted for
western blotting to assess the levels of Smad4, MDR1, Bcl-2, Bax
and caspase-3, as well as the phosphorylation of NF-κB p65.
Combination for subcutaneous tumor
inhibition
Six-week-old male BALB/c nude mice, provided by the
Animal Experiment Center of the Second Military Medical University,
were randomly divided into 3 groups: the model group (16 mice,
inoculated with Caco-2/PT), the Lv-control group (8 mice,
inoculated with Caco-2/PT infected with Lv-control) and the Smad4
overexpression group (8 mice, inoculated with Caco-2/PT infected
with Lv-Smad4). Suspensions of Caco-2/PT, Caco-2/PT + Lv-control or
Caco-2/PT + Lv-Smad4 (1×107 cells) cells were prepared
in 50 µl of dPBS and subcutaneously injected into the left
fore-flank of each mouse; tumors formed within 15 days. The model
group was divided into treated and untreated groups. The treated,
Lv-control and Smad4 overexpression groups were administered daily
with 10 mg/kg PT [200 ng/µl, prepared in normal saline from a 20
µg/µl stock solution prepared with dimethyl sulfoxide (DMSO)] via
caudal vein injection, and the untreated group was injected with
normal saline. The treatment period was 5 weeks. The tumor diameter
was assessed every week, and the tumor volumes were calculated. All
mouse experiments were approved by the Medical Ethics Review
Committee of Harbin Medical University.
Assessment of mRNA levels
Total RNA was isolated using TRIzol reagent
(Invitrogen) according to the manufacturer's instruction and
reverse-transcribed into cDNA using M-MLV reverse transcriptase and
an oligo(dT)18 primer (both from Takara, Shiga, Japan). The
following specific primers were used for quantitative PCR of human
Smad4 and β-actin to generate amplified products of 222 and 211 bp,
respectively: Smad4, 5′-ACCACCAAAACGGCCATCTTCAG-3′ and
5′-GGTCCACGTATCCATCAACAGTA-3′; β-actin, 5′-CCTGTACGCCAACACAGTGC-3′
and 5′-ATACTCCTGCTTGCATCC-3′. Real-time PCR was performed using a
SYBR® Premix Ex Taq™ kit and a TP800 system (both from
Takara). cDNA from 200 ng of total RNA was used as the template.
The PCR reactions were carried out under the following conditions:
40 cycles of denaturation at 95°C for 10 sec, annealing at 60°C for
20 sec and extension at 72°C for 20 sec. The mRNA levels of Smad4
were normalized to those of an endogenous housekeeping gene,
β-actin, using the ΔΔCt method.
Assessment of protein levels
Total protein was extracted from cells using M-PER
mammalian protein extraction reagent (Pierce, Rockford, IL, USA).
Equal amounts of protein (25 µg/lane), as estimated using the
bicinchoninic acid (BCA) protein assay (Pierce), were separated by
11% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto nitrocellulose membranes. The blots
were probed with a monoclonal antibody against human Smad4
(1:1,000), anti-MDR1 (1:500), anti-p-p65/p65 (1:500), anti-Bcl-2
(1:200), anti-Bax (1:500), anti-caspase-3 (1:300) and anti-β-actin
(1:800), followed by a secondary horseradish peroxidase
(HRP)-conjugated anti-mouse/rabbit antibody (Santa Cruz
Biotechnology, Santa Cruz, CA, USA). After being washed, the bands
were detected using chemiluminescence and exposed to X-ray films.
β-actin was used as an endogenous reference for normalization.
Statistical analysis
All data are expressed as the mean ± standard
deviation (SD). Factorial analysis was employed for inter- and
intra-group comparisons. Statistical significance was determined by
Student's t-test for comparison of two groups, and means were
compared with a Chi-square test. P-values <0.05 were considered
to be statistically significant. All statistical analyses were
performed using SPSS 13.0 software (SPSS, Inc., Chicago, IL,
USA).
Results
Assessment of Smad4 mRNA and
protein
The real-time PCR and western blotting results for
Smad4 in 3 PT-resistant cell lines revealed decreases at both the
transcriptional level (Fig. 1A-a, B-a
and C-a) and protein level (Fig.
1A-b, B-b and C-b). There was a significant difference in
PT-resistant cells and parental cells (P<0.01).
Lentiviral-mediated overexpression of Smad4 in these 3 PT-resistant
cell lines resulted in a gene delivery efficiency close to 100% at
72 h after infection, as demonstrated by green fluorescent protein
(GFP) expression (Fig. 1A-c, B-c and
C-c). Furthermore, protein assessments revealed that the
protein levels of Smad4 (Fig. 1A-d, B-d
and C-d) were significantly increased in the groups infected
with Lv-Smad4 compared with the control cells or the Lv-control
group (P<0.01), and there was no significant difference in the
control cells or the Lv-control group (P>0.05), indicating that
the lentiviral system effectively delivered exogenous Smad4 gene in
these cells.
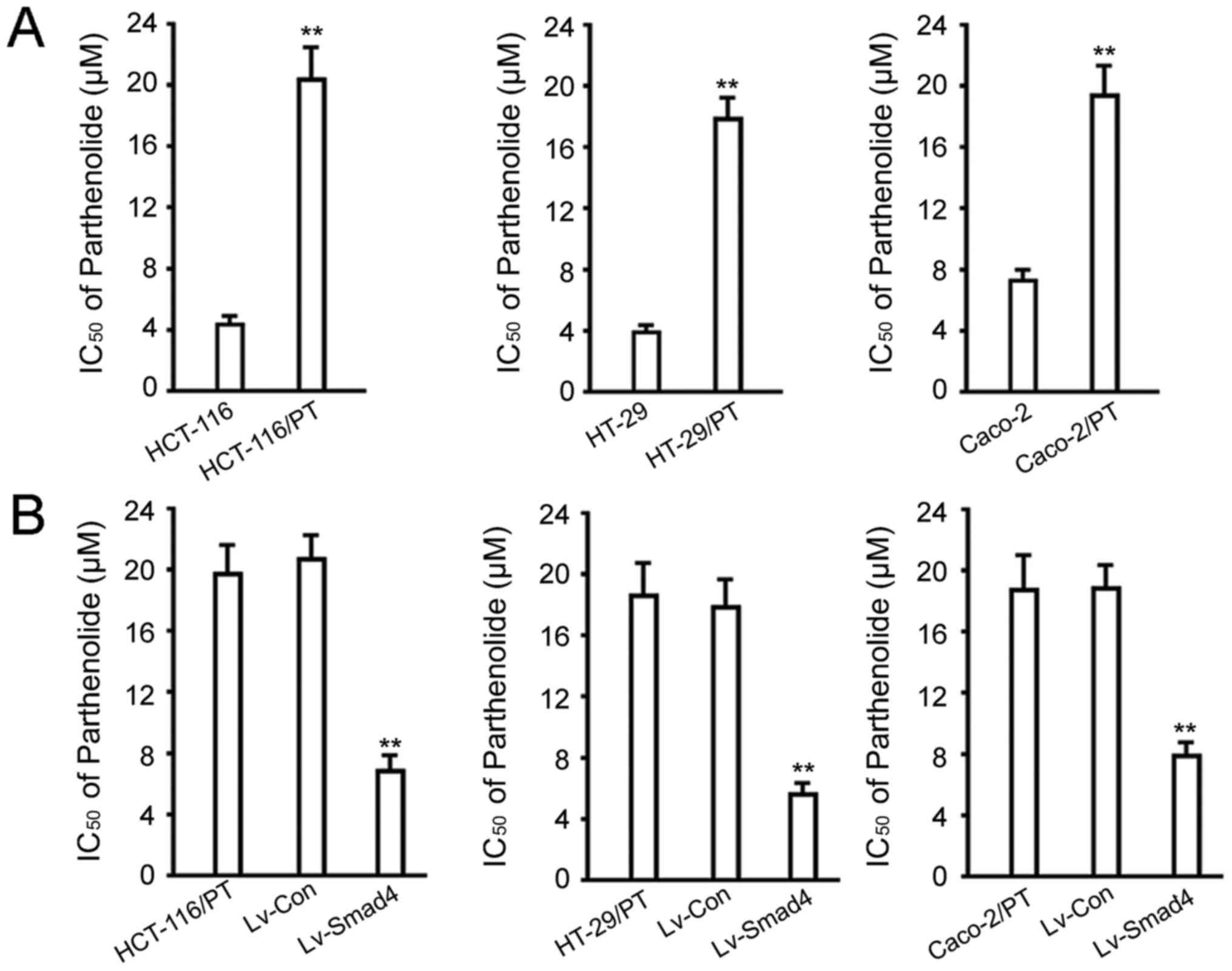
IC50 value analysis
We analyzed the IC50 values to ascertain
the successful establishment of PT-resistant cell lines. As
expected, the IC50 values of the HCT-116/PT, HT-29/PT
and Caco-2/PT cell lines increased from 4.09±0.76, 3.97±0.58 and
7.13±1.21 µM to 20.31±3.21, 17.65±1.89 and 18.64±3.02 µM after 48
h, respectively (P<0.01, vs. the parental cells; Fig. 2A). Overexpression of Smad4 in
HCT-116/PT, HT-29/PT and Caco-2/PT cells significantly decreased
the IC50 values to 6.12±2.11, 5.75±1.08 and 7.86±1.95 µM
(P<0.01, vs. the control cells or the Lv-control group; Fig. 2B), respectively.
Assessment of cell migration and
apoptosis
We employed a Transwell experiment to explore
changes in tumor cell migration, which plays an important role in
tumor metastasis. In the absence of PT, there was no statistical
difference in the number of cells penetrating the basal membrane
between the Lv-Smad4 and the control cells or the Lv-control group
(P>0.05); in the presence of 10 µM PT, overexpression of Smad4
decreased the number of cells that penetrated the basement membrane
(P<0.01, vs. the control cells or the Lv-control group; Fig. 3A), and there was no statistical
difference between the control cell group and Lv-control group
(P>0.05). The data also revealed that treatment with 10 µM PT
had no significant impact on HT-29 and HT-29 infected with
Lv-control (P>0.05), however it significantly inhibited the
migration in HT-29 cells infected with Lv-Smad4 (P<0.05, PT
group vs. non-PT group;Fig. 3A).
The results indicate that overexpression of Smad4 has no direct
impact on the migration of HT-29/PT, but may increase its
sensitivity to PT, thus an increased response to PT was observed.
Consistent with the data of the migration assay, overexpression of
Smad4 did not induce apoptosis in HT-29/PT cells, but it restored
its sensitivity to PT, thus PT exerts its apoptosis-inducing
activity (Fig. 3B). We also
performed these assays in HCT-116/PT and Caco-2/PT cell lines, and
observed similar results (data not shown).
Tumor inhibition experiment
The results of the in vivo tumor inhibition
experiment revealed that administration of PT for 5 weeks only
slightly inhibited tumor formation by Caco-2/PT cells, but
significantly inhibited tumor formation by Caco-2/PT cells
overexpressing Smad4 (Fig. 4). At
the fifth week after administration, the tumor volumes were
535.29±80.29 mm3 in the model group and 431.40±60.00
mm3 in the PT administration group, corresponding to an
inhibition rate of 19.42% and a non-significant difference.
However, the tumor volume in the Smad4-overexpressing group that
received PT averaged 189.75±42.18 mm3, corresponding to
an inhibition rate of 64.55% and a significant difference compared
to the control group (P<0.01). The data from the in vivo
tumor inhibition experiment were in line with the overexpression of
Smad4 resensitizing the 3 PT-resistant cell lines.
Analysis of the pathway involved in
Smad4 overexpression-mediated increases in sensitivity to PT in
resistant colorectal cancer cells
To elucidate how Smad4 overexpression increases the
sensitivity of resistant colorectal cancer cells to PT, we examined
a number of proteins that are related to Smad4 expression and are
associated with drug resistance. In the absence of PT, the level of
Smad4 in the Lv-Smad4 group was significantly higher than that in
the control cells (P<0.01; Fig.
5). The opposite was observed with MDR1. No observable
difference was found in the phosphorylation of NF-κB p65, and in
the expression of Bcl-2, caspase-3 and Bax (P>0.05). When the
cells were treated with 10 µM PT for 48 h, caspase-3 and Bax in the
cells infected with Lv-Smad4 were higher than in the control group
(P<0.01), and the expression of MDR1 and Bcl-2 and the
phosphorylation of NF-κB p65 in the Lv-Smad4 group were lower than
in the control group (P<0.05). The data of HCT-116/PT and
HT-29/PT cell lines were in agreement with those of Caco-2/PT (data
not shown).
Discussion
Recent studies have established the antitumor role
of parthenolide (PT). For example, Kim et al demonstrated
that PT decreases NF-κB p65 expression by blocking phosphorylation
and subsequently degrading inhibitor of κB-α (IκBα). PT also
promotes apoptosis by downregulating Bcl-2 and Bcl-xL, two
anti-apoptosis proteins, suggesting a potential therapeutic role of
PT in colitis-associated colon cancer (12). Zhao et al described that PT
not only triggers extrinsic apoptosis by upregulating TNFRSF10B and
downregulating CFLAR, but also induces intrinsic apoptosis by
increasing the expression of PMAIP1 and decreasing the level of
MCL1 in non-small cell lung cancer (NSCLC) cells (13). In summary, these studies revealed
that PT has an anticancer role that should not be ignored. Our
analyses revealed that although PT inhibited proliferation in
colorectal cancer cells, drug resistance developed over the course
of administration, which were accompanied by changes in protein
expression. The study of the relationship between PT and drug
resistance has primarily focused on two aspects: i) the ability of
PT to directly inhibit the proliferation of tumor cells and ii) the
ability of PT to increase chemosensitivity or reverse resistance to
chemotherapeutic agents. Specifically, Ghantous et al
demonstrated that PT inhibits JB6P+ cell proliferation
by suppressing tumor promoter-mediated NF-кB activation (14). Furthermore, Li et al
demonstrated that PT decreased levels of MMP-9, VEGF and IL-8
expression in breast cancer cells and inhibited the proliferation
and migration of MDA-MB-231 cells as well as the formation of
lumen-like structures by human umbilical vein endothelial cells
(15). Alternatively, PT has been
reported to inhibit the activity of NF-кB and cell growth in 3
gastric cancer cell lines and to increase chemosensitivity
(16). In particular, PT enhanced
doxorubicin (DOX) aggregation and the apoptotic cytotoxicity of DOX
in A549-derived doxorubicin (A549/DOX)-resistant cells and reversed
DOX resistance by suppressing p-gp expression via mechanisms that
involve attenuation of NF-κB activation and HSP70 upregulation
(17). Furthermore, parthenolide
significantly increased 5-FU-mediated inhibition of BEL-7402/5-FU
cell (hepatic carcinoma multidrug-resistant cells) proliferation
and reversed the resistance of hepatic carcinoma-resistant cells
(18). However, resistance to PT
itself has not yet been reported. When treating colorectal cancer
cells with PT, we found that long-term treatment induced
resistance. Thus, we investigated the development of this
resistance in colorectal cancer cell lines by examining the key
proteins involved and strategies for reversing this resistance.
Smad4, which is located in the 18q21 region
(19), is a tumor suppressor that
is frequently inactivated in tumors (20). Indeed, mutation or inactivation of
Smad4 not only results in a lack of TGF-β1-induced growth
inhibition (21), but also affects
the chemosensitivity of cancer cells (22,23).
Moreover, Smad4 deficiency induced chemoresistance to 5-FU in CT26
and SW620 cells both in vitro and in vivo.
Furthermore, Smad4 deficiency increased resistance to 5-FU by
attenuating G1 or G2 cell cycle arrest via the
PI3K/Akt/CDC2/survivin pathway activation (24). Yu et al revealed that HDAC4
regulates Smad4 expression by inducing histone H3 deacetylation in
the Smad4 promoter region. These authors also demonstrated that
histone deacetylase 4 (HDAC4)-mediated deacetylation of the Smad4
promoter may lead to 5-FU resistance in breast cancer cells
(25). These studies all revealed
that Smad4 influences the response of tumor cells to 5-FU via
different pathways. However, the effect of Smad4 on the sensitivity
of tumor cells to PT has not been reported to date.
With the advance in clinical chemotherapy of
colorectal cancer, many new chemotherapeutic agents have been
researched, including PT (17). The
present study revealed that although PT inhibited colon cancer, the
cells easily developed resistance. Moreover, we revealed that Smad4
mRNA and protein levels differed between resistant cells and
parental cells. Accordingly, we examined the mechanism by which
Smad4 expression affected the response of colorectal cancer cell
lines to PT. To this end, we employed a lentivirus to overexpress
Smad4 in resistant cells, which decreased the IC50
values of PT in colorectal cancer cell lines. Specifically, PT
significantly inhibited the migration and in vivo growth of
these tumor cells and promoted their apoptosis, suggesting that
Smad4 expression affects the chemosensitivity of colorectal cancer
cell lines to PT. Therefore, combined interventions based on Smad4
and PT may produce a better outcome than application of PT alone to
PT-resistant colorectal cancer. How does Smad4 mediate this effect?
For example, increases in MDR1, an ATP-dependent membrane transport
protein involved in drug resistance, reportedly decreased
accumulation of anticancer drugs in cells (26). However, we found that Smad4
decreased the levels of this protein. Based on these data, we
concluded that in these 3 PT-resistant cell lines, Smad4
overexpression directly downregulated the expression of the the
MDR1 protein, and consequently resulted in the accumulation of PT,
which inhibited the phosphorylation of NF-κB p65 and the expression
of Bcl-2, and promoted the expression of Bax and caspase-3. All
alterations of these proteins associated with cell migration and
apoptosis are consistent with previous studies on the mechanism
through which PT suppresses tumor cells. Therefore, the
overexpression of Smad4 may increase PT accumulation in resistant
cells by suppressing MDR1 and significantly decreasing
IC50 values in these resistant cell lines. Based on
these findings, the following questions warrant further
investigation in the near future: Why is Smad4 expression
suppressed in PT-resistant colorectal cancer cells? What is the
target through which Smad4 restores the sensitivity to PT in
resistant colorectal cancer cells? And how does the change in Smad4
expression dynamically affect the content of PT in the resistant
cells?
In summary, drug resistance significantly influenced
the survival of patients with cancer. The present study revealed
that Smad4 re-expression may be crucial for reversing the
resistance to PT in PT-resistant colorectal cancer.
References
|
1
|
Sun J, Zhang C, Bao YL, Wu Y, Chen ZL, Yu
CL, Huang YX, Sun Y, Zheng LH, Wang X, et al: Parthenolide-induced
apoptosis, autophagy and suppression of proliferation in HepG2
cells. Asian Pac J Cancer Prev. 15:4897–4902. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carlisi D, D'Anneo A, Martinez R, Emanuele
S, Buttitta G, Di Fiore R, Vento R, Tesoriere G and Lauricella M:
The oxygen radicals involved in the toxicity induced by
parthenolide in MDA-MB-231 cells. Oncol Rep. 32:167–172. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Al-Fatlawi AA, Al-Fatlawi AA, Irshad M,
Rahisuddin and Ahmad A: Effect of parthenolide on growth and
apoptosis regulatory genes of human cancer cell lines. Pharm Biol.
53:104–109. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu C, Wang W, Jia Y, Liu X, Tong Z and Li
B: Inhibition of AMPK/autophagy potentiates parthenolide-induced
apoptosis in human breast cancer cells. J Cell Biochem.
115:1458–1466. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu HJ, Jung JY, Jeong JH, Cho SD and Lee
JS: Induction of apoptosis by parthenolide in human oral cancer
cell lines and tumor xenografts. Oral Oncol. 51:602–609. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang
J, Perry SR, Labrot ES, Wu X, Lis R, et al: SMAD4-dependent barrier
constrains prostate cancer growth and metastatic progression.
Nature. 470:269–273. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen C, Sun MZ, Liu S, Yeh D, Yu L, Song
Y, Gong L, Hao L, Hu J and Shao S: Smad4 mediates malignant
behaviors of human ovarian carcinoma cell through the effect on
expressions of E-cadherin, plasminogen activator inhibitor-1 and
VEGF. BMB Rep. 43:554–560. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inamoto S, Itatani Y, Yamamoto T,
Minamiguchi S, Hirai H, Iwamoto M, Hasegawa S, Taketo MM, Sakai Y
and Kawada K: Loss of SMAD4 promotes colorectal cancer progression
by accumulation of myeloid-derived suppressor cells through the
CCL15-CCR1 chemokine axis. Clin Cancer Res. 22:492–501. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nikolic A, Ristanovic M, Zivaljevic V,
Rankov AD, Radojkovic D and Paunovic I: SMAD4 gene promoter
mutations in patients with thyroid tumors. Exp Mol Pathol.
99:100–103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boulay JL, Mild G, Lowy A, Reuter J,
Lagrange M, Terracciano L, Laffer U, Herrmann R and Rochlitz C:
SMAD4 is a predictive marker for 5-fluorouracil-based chemotherapy
in patients with colorectal cancer. Br J Cancer. 87:630–634. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Papageorgis P, Cheng K, Ozturk S, Gong Y,
Lambert AW, Abdolmaleky HM, Zhou JR and Thiagalingam S: Smad4
inactivation promotes malignancy and drug resistance of colon
cancer. Cancer Res. 71:998–1008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim SL, Liu YC, Seo SY, Kim SH, Kim IH,
Lee SO, Lee ST, Kim DG and Kim SW: Parthenolide induces apoptosis
in colitis-associated colon cancer, inhibiting NF-κB signaling.
Oncol Lett. 9:2135–2142. 2015.PubMed/NCBI
|
|
13
|
Zhao X, Liu X and Su L: Parthenolide
induces apoptosis via TNFRSF10B and PMAIP1 pathways in human lung
cancer cells. J Exp Clin Cancer Res. 33:32014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ghantous A, Saikali M, Rau T,
Gali-Muhtasib H, Schneider-Stock R and Darwiche N: Inhibition of
tumor promotion by parthenolide: Epigenetic modulation of
p21. Cancer Prev Res. 5:1298–1309. 2012. View Article : Google Scholar
|
|
15
|
Li CJ, Guo SF and Shi TM: Culture
supernatants of breast cancer cell line MDA-MB-231 treated with
parthenolide inhibit the proliferation, migration, and lumen
formation capacity of human umbilical vein endothelial cells. Chin
Med J. 125:2195–2199. 2012.PubMed/NCBI
|
|
16
|
Sohma I, Fujiwara Y, Sugita Y, Yoshioka A,
Shirakawa M, Moon JH, Takiguchi S, Miyata H, Yamasaki M, Mori M, et
al: Parthenolide, an NF-κB inhibitor, suppresses tumor growth and
enhances response to chemotherapy in gastric cancer. Cancer
Genomics Proteomics. 8:39–47. 2011.PubMed/NCBI
|
|
17
|
Xin Y, Yin F, Qi S, Shen L, Xu Y, Luo L,
Lan L and Yin Z: Parthenolide reverses doxorubicin resistance in
human lung carcinoma A549 cells by attenuating NF-κB activation and
HSP70 up-regulation. Toxicol Lett. 221:73–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu D, Liu Y, Liu M, Ran L and Li Y:
Reversing resistance of multidrug-resistant hepatic carcinoma cells
with parthenolide. Future Oncol. 9:595–604. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ramachandra M, Atencio I, Rahman A,
Vaillancourt M, Zou A, Avanzini J, Wills K, Bookstein R and Shabram
P: Restoration of transforming growth factor Beta signaling by
functional expression of smad4 induces anoikis. Cancer Res.
62:6045–6051. 2002.PubMed/NCBI
|
|
20
|
Li X, Liu B, Xiao J, Yuan Y, Ma J and
Zhang Y: Roles of VEGF-C and Smad4 in the lymphangiogenesis,
lymphatic metastasis, and prognosis in colon cancer. J Gastrointest
Surg. 15:2001–2010. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bierie B and Moses HL: Tumour
microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer.
Nat Rev Cancer. 6:506–520. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kozak MM, von Eyben R, Pai J, Vossler SR,
Limaye M, Jayachandran P, Anderson EM, Shaffer JL, Longacre T, Pai
RK, et al: Smad4 inactivation predicts for worse prognosis and
response to fluorouracil-based treatment in colorectal cancer. J
Clin Pathol. 68:341–345. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alhopuro P, Alazzouzi H, Sammalkorpi H,
Dávalos V, Salovaara R, Hemminki A, Järvinen H, Mecklin JP,
Schwartz S Jr, Aaltonen LA, et al: SMAD4 levels and response to
5-fluorouracil in colorectal cancer. Clin Cancer Res. 11:6311–6316.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang B, Leng C, Wu C, Zhang Z, Dou L, Luo
X, Zhang B and Chen X: Smad4 sensitizes colorectal cancer to
5-fluorouracil through cell cycle arrest by inhibiting the
PI3K/Akt/CDC2/survivin cascade. Oncol Rep. 35:1807–1815. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu SL, Lee DC, Son JW, Park CG, Lee HY and
Kang J: Histone deacetylase 4 mediates SMAD family member 4
deacetylation and induces 5-fluorouracil resistance in breast
cancer cells. Oncol Rep. 30:1293–1300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abdallah HM, Al-Abd AM, El-Dine RS and
El-Halawany AM: P-glycoprotein inhibitors of natural origin as
potential tumor chemo-sensitizers: A review. J Adv Res. 6:45–62.
2015. View Article : Google Scholar : PubMed/NCBI
|